SPATA & The tidyverse
spata-in-the-tidyverse.RmdThe tidyverse is a collection of R-packages that provide consistent tools to work with data.frames in a tidy-data fashion. SPATA attempts to provide it’s results in a way that you can leverage the power of these tools. The following two examples are supposed to give insight in our approaches and to highlight the use of SPATA’s joinWith-family in combination with the tidyverse.
Example 1: Four states
Neftel et al. 2019 (Figure 3) suggested an integrative model which assigned glioblastoma-cells into four different states according to the results of the conducted gene expression analysis. The distribution of these states was displayed in a scatterplot in which each dot represented a cell and the dot’s position represented it’s differentiation level towards these defined states. The concept of this visualization technique is manifested in the SPATA-function plotFourStates() and can be emulated with just a handful of tidyverse-functions.
#load packages library(SPATA) library(tidyverse) library(magrittr) # load glioblastoma sample spata_obj <- loadSpataObject("data/spata-obj-four-states-example.RDS") # the respective states were stored in the gene-set data.frame via addGeneSets() neftel_states <- c("Neftel_OPClike","Neftel_NPClike2", "Neftel_AClike", "Neftel_Mes_Comb") # output neftel_states
## [1] "Neftel_OPClike" "Neftel_NPClike2" "Neftel_AClike" "Neftel_Mes_Comb"# display the respective genes getGenes(object = spata_obj, of_gene_sets = neftel_states, simplify = FALSE)
## $Neftel_OPClike
## [1] "BCAN" "PLP1" "GPR17" "FIBIN" "LHFPL3" "OLIG1" "PSAT1"
## [8] "SCRG1" "OMG" "APOD" "SIRT2" "TNR" "THY1" "PHYHIPL"
## [15] "NKAIN4" "PTPRZ1" "VCAN" "DBI" "PMP2" "CNP" "TNS3"
## [22] "LIMA1" "CA10" "PCDHGC3" "CNTN1" "SCD5" "P2RX7" "CADM2"
## [29] "TTYH1" "FGF12" "TMEM206" "NEU4" "FXYD6" "RNF13" "RTKN"
## [36] "GPM6B" "LMF1" "ALCAM" "PGRMC1" "HRASLS" "BCAS1" "RAB31"
## [43] "PLLP" "FABP5" "NLGN3" "SERINC5" "EPB41L2" "GPR37L1"
##
## $Neftel_NPClike2
## [1] "STMN2" "CD24" "RND3" "TUBB3" "MIAT"
## [6] "DCX" "NSG1" "ELAVL4" "MLLT11" "DLX6-AS1"
## [11] "SOX11" "NREP" "FNBP1L" "TAGLN3" "STMN4"
## [16] "DLX5" "SOX4" "MAP1B" "RBFOX2" "STMN1"
## [21] "TMEM161B-AS1" "DPYSL3" "SEPT3" "PKIA" "ATP1B1"
## [26] "DYNC1I1" "CD200" "SNAP25" "PAK3" "NDRG4"
## [31] "KIF5A" "UCHL1" "ENO2" "KIF5C" "DDAH2"
## [36] "TUBB2A" "LBH" "TCF4" "GNG3" "NFIB"
## [41] "DPYSL5" "CRABP1" "DBN1" "NFIX" "CEP170"
## [46] "BLCAP"
##
## $Neftel_AClike
## [1] "CST3" "S100B" "SLC1A3" "HEPN1" "HOPX" "MT3" "SPARCL1"
## [8] "MLC1" "GFAP" "FABP7" "BCAN" "PON2" "METTL7B" "SPARC"
## [15] "GATM" "RAMP1" "PMP2" "AQP4" "DBI" "EDNRB" "PTPRZ1"
## [22] "CLU" "PMP22" "ATP1A2" "S100A16" "HEY1" "PCDHGC3" "TTYH1"
## [29] "NDRG2" "PRCP" "ATP1B2" "AGT" "PLTP" "GPM6B" "F3"
## [36] "RAB31" "ANXA5" "TSPAN7"
##
## $Neftel_Mes_Comb
## [1] "HILPDA" "ADM" "DDIT3" "NDRG1" "HERPUD1" "DNAJB9"
## [7] "TRIB3" "ENO2" "AKAP12" "SQSTM1" "MT1X" "NAMPT"
## [13] "NRN1" "SLC2A1" "BNIP3" "LGALS3" "INSIG2" "IGFBP3"
## [19] "PPP1R15A" "VIM" "PLOD2" "GBE1" "SLC2A3" "FTL"
## [25] "WARS" "XPOT" "HSPA5" "GDF15" "ANXA2" "EPAS1"
## [31] "LDHA" "P4HA1" "SERTAD1" "PFKP" "PGK1" "EGLN3"
## [37] "SLC6A6" "CA9" "BNIP3L" "TRAM1" "UFM1" "ASNS"
## [43] "GOLT1B" "ANGPTL4" "SLC39A14" "CDKN1A" "HSPA9" "CHI3L1"
## [49] "ANXA2" "ANXA1" "CD44" "VIM" "MT2A" "C1S"
## [55] "NAMPT" "EFEMP1" "C1R" "SOD2" "IFITM3" "TIMP1"
## [61] "SPP1" "A2M" "S100A11" "MT1X" "S100A10" "FN1"
## [67] "LGALS1" "S100A16" "CLIC1" "MGST1" "RCAN1" "TAGLN2"
## [73] "NPC2" "SERPING1" "EMP1" "APOE" "CTSB" "C3"
## [79] "LGALS3" "MT1E" "EMP3" "SERPINA3" "ACTN1" "PRDX6"
## [85] "IGFBP7" "SERPINE1" "PLP2" "MGP" "CLIC4" "GFPT2"
## [91] "GSN" "NNMT" "TUBA1C" "GJA1" "TNFRSF1A" "WWTR1"# display the states on the surface plotSurfaceComparison(object = spata_obj, variables = neftel_states, smooth = TRUE, display_image = TRUE, pt_alpha = 0.5, pt_size = 1.5)
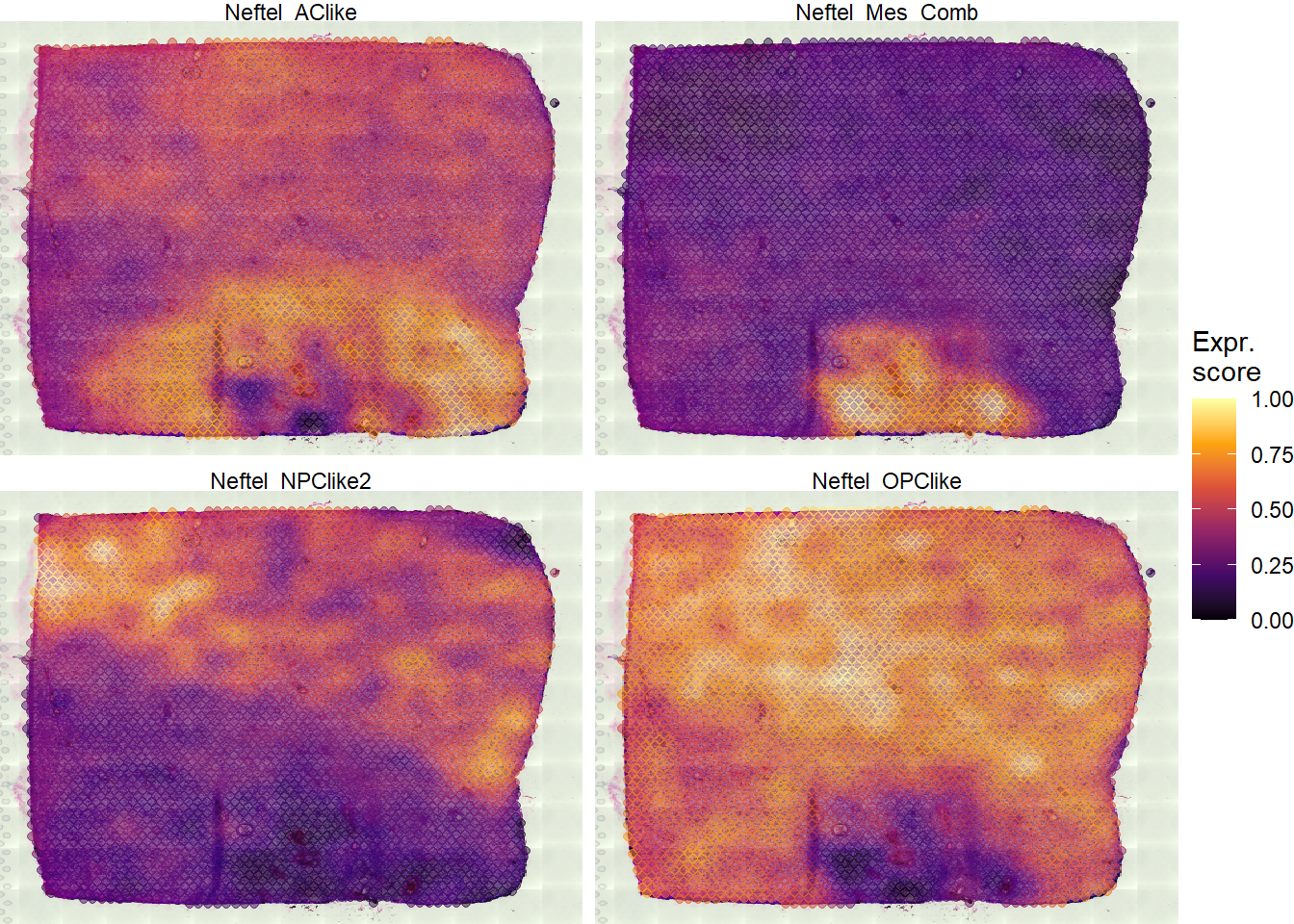
Figure 1.1 Surface plots color according to each of the four cellular states of Neftel et al. 2019.
1. Assemble the appropriate data.frame
neftel_df <- joinWithGeneSets(object = spata_obj, spata_df = getSpataDf(spata_obj), gene_sets = neftel_states) # output neftel_df
2. Compute the respective position of every barcode spot
# 1. shift the data.frame shifted_df <- tidyr::pivot_longer( data = neftel_df, cols = all_of(neftel_states), names_to = "gene_set", values_to = "gene_set_expr" ) # output shifted_df
# 2. figure out which of the four states is a barcode's maximum # by filtering for it max_localisation <- group_by(shifted_df, barcodes) %>% filter(gene_set_expr == max(gene_set_expr)) %>% ungroup() %>% # rename the remaining gene sets to 'max_gene_set' select(barcodes, max_gene_set = gene_set, max_expr = gene_set_expr) %>% # assign the vertical localization of the state plot depending on where the maximum occured with a dichotome string mutate(max_loc = if_else(max_gene_set %in% neftel_states[1:2], true = "top", false = "bottom")) # output max_localisation
# 3. calculate the x-position with_x_positions <- # rejoin the data left_join(x = neftel_df, y = max_localisation, by = "barcodes") %>% # calculate the position on the x-axis by comparing the respective gene-set expressions mutate( pos_x = case_when( max_loc == "top" & Neftel_OPClike > Neftel_NPClike2 ~ (log2(abs((Neftel_OPClike - Neftel_NPClike2) + 1)) * -1), max_loc == "top" & Neftel_NPClike2 > Neftel_OPClike ~ log2(abs((Neftel_NPClike2 - Neftel_OPClike) + 1)), max_loc == "bottom" & Neftel_AClike > Neftel_Mes_Comb ~ (log2(abs((Neftel_AClike - Neftel_Mes_Comb) + 1)) * -1), max_loc == "bottom" & Neftel_Mes_Comb > Neftel_AClike ~ log2(abs((Neftel_Mes_Comb - Neftel_AClike) + 1))) ) # output with_x_positions
# 4. calculate the y-position final_df <- group_by(.data = with_x_positions, barcodes) %>% mutate( pos_y = case_when( max_loc == "bottom" ~ (log2(abs(max(c(Neftel_AClike, Neftel_Mes_Comb)) - max(Neftel_OPClike, Neftel_NPClike2) + 1)) * -1), max_loc == "top" ~ log2(abs(max(c(Neftel_OPClike, Neftel_NPClike2)) - max(Neftel_AClike, Neftel_Mes_Comb) + 1)) ) ) %>% # filter out na values filter(!is.na(pos_x) & !is.na(pos_y)) # output final_df
3. Plotting with ggplot2
Once the data-wrangling part is done we can plot the data. Note that the output data.frame is still a tidy spata-data.frame. To display the seurat_clusters-feauture as well we need to join it to our data.
final_df <- joinWith(spata_obj, spata_df = final_df, features = "seurat_clusters") max <- max(abs(final_df$pos_x), abs(final_df$pos_y)) ggplot(data = final_df) + geom_vline(xintercept = 0, linetype = "dashed", color = "lightgrey") + geom_hline(yintercept = 0, linetype = "dashed", color = "lightgrey") + # map the calculated positions on the x- and y-aesthetic geom_point(mapping = aes(x = pos_x, y = pos_y, color = seurat_clusters, shape = max_gene_set), size = 1.5, alpha = 0.75, data = final_df) + scale_x_continuous(limits = c(-max*1.1, max*1.1), expand = c(0,0)) + scale_y_continuous(limits = c(-max*1.1, max*1.1), expand = c(0,0)) + scale_color_add_on(clrp = "npg", variable = "discrete") + theme_bw() + theme( panel.grid.major = element_blank(), panel.grid.minor = element_blank() ) + labs(x = NULL, y = NULL, color = "Seurat Clusters", shape = "Max. Expr.")
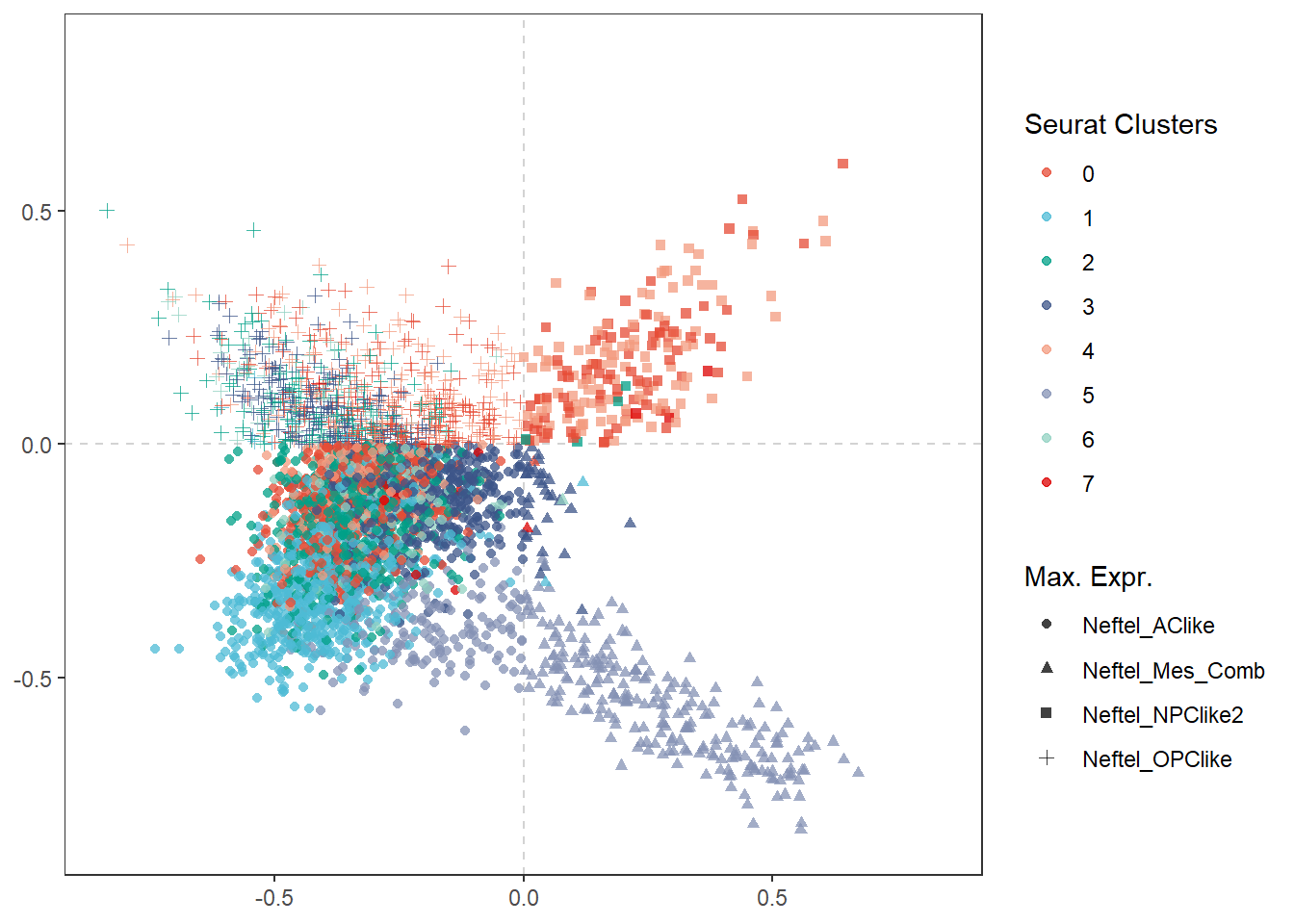
Figure 1.2 The barcode spots relocated according to their differentation.
# Surface plot in comparison (compare with Figure 1.1) plotSurface(object = spata_obj, color_to = "seurat_clusters", pt_clrp = "npg")
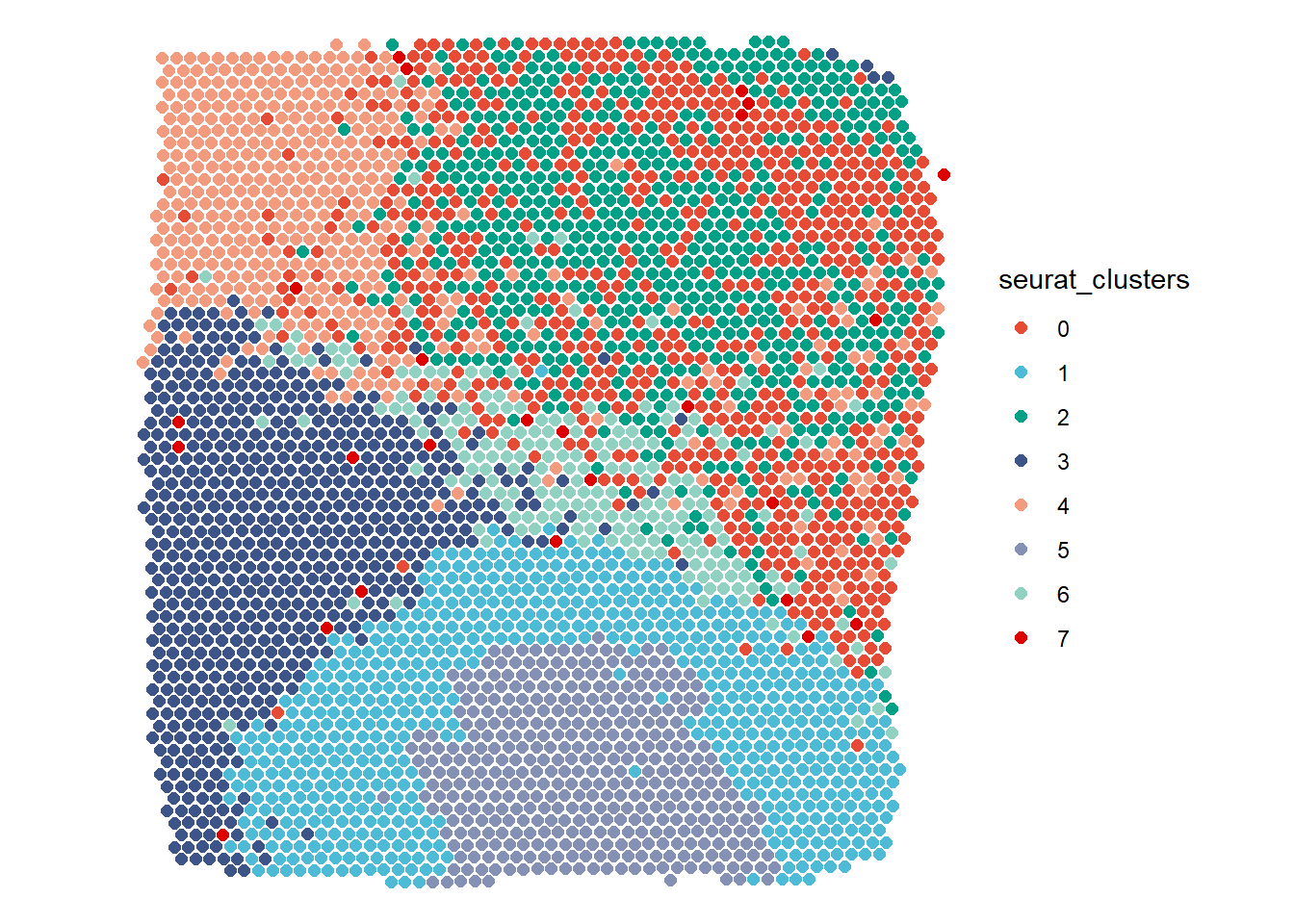
Figure 1.2 The barcode spots relocated according to their differentation.
Example 2 - Handling Spatial Trajectories
The loaded spata-object contains spatial-expression data about a brain slice going through all 6 cortical layers of the central nervous system (CNS-layers). Additionally it contains a spatial trajectory that we have drawn with createTrajectories() through all those layers and therefore called cns-layers. According to Seurat’s clustering algorithm the barcode-spots cluster together in an expected way - representing the CNS-layers - as shown in the figures below.
plotSurface(spata_obj_cns, color_to = "seurat_clusters", pt_clrp = "npg") plotTrajectory(spata_obj_cns, trajectory_name = "cns-layers", color_to = "seurat_clusters", display_image = FALSE, pt_alpha = 0.4, pt_clrp = "npg", sgmt_size = 1.5) + theme(legend.position = "none")
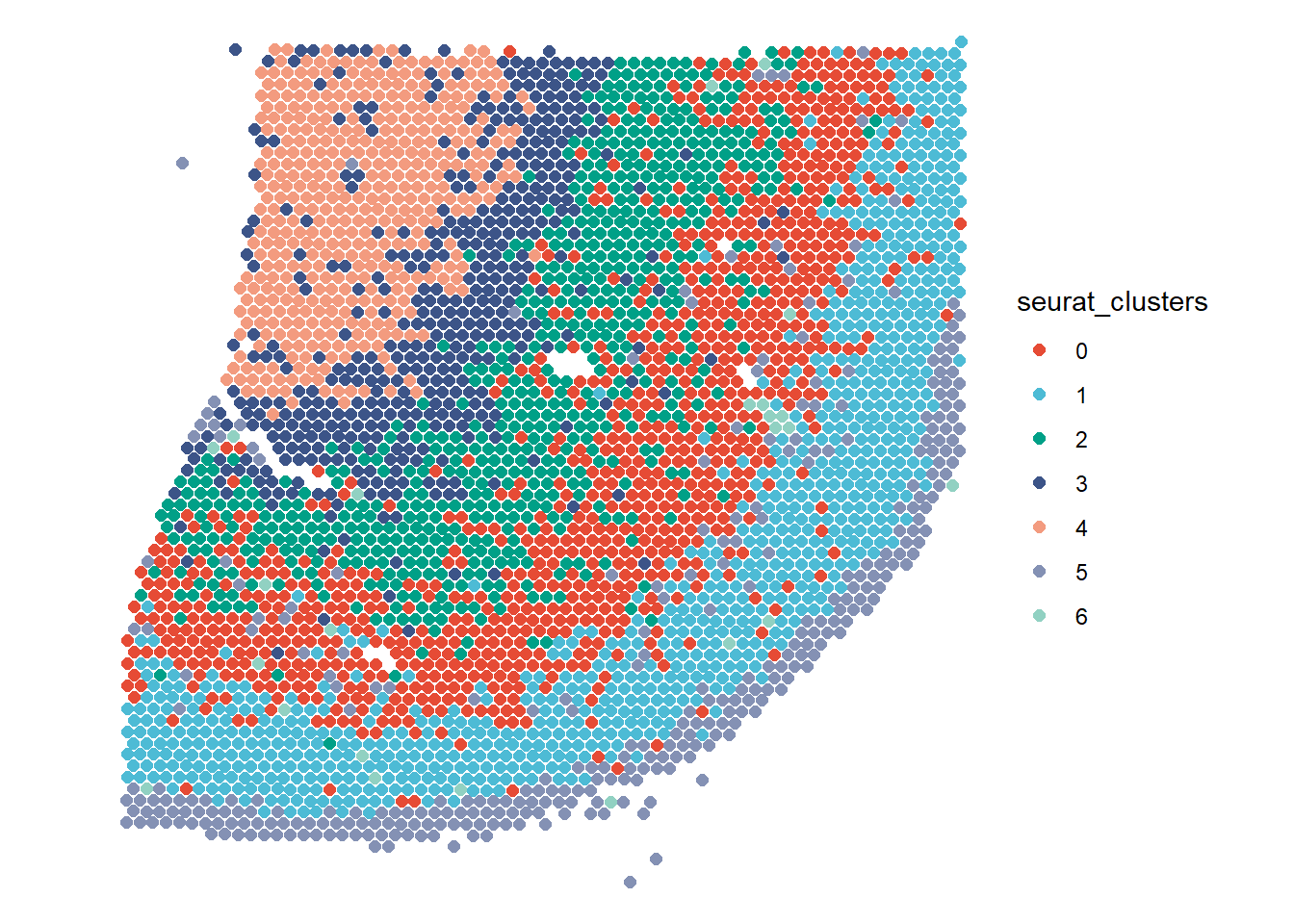
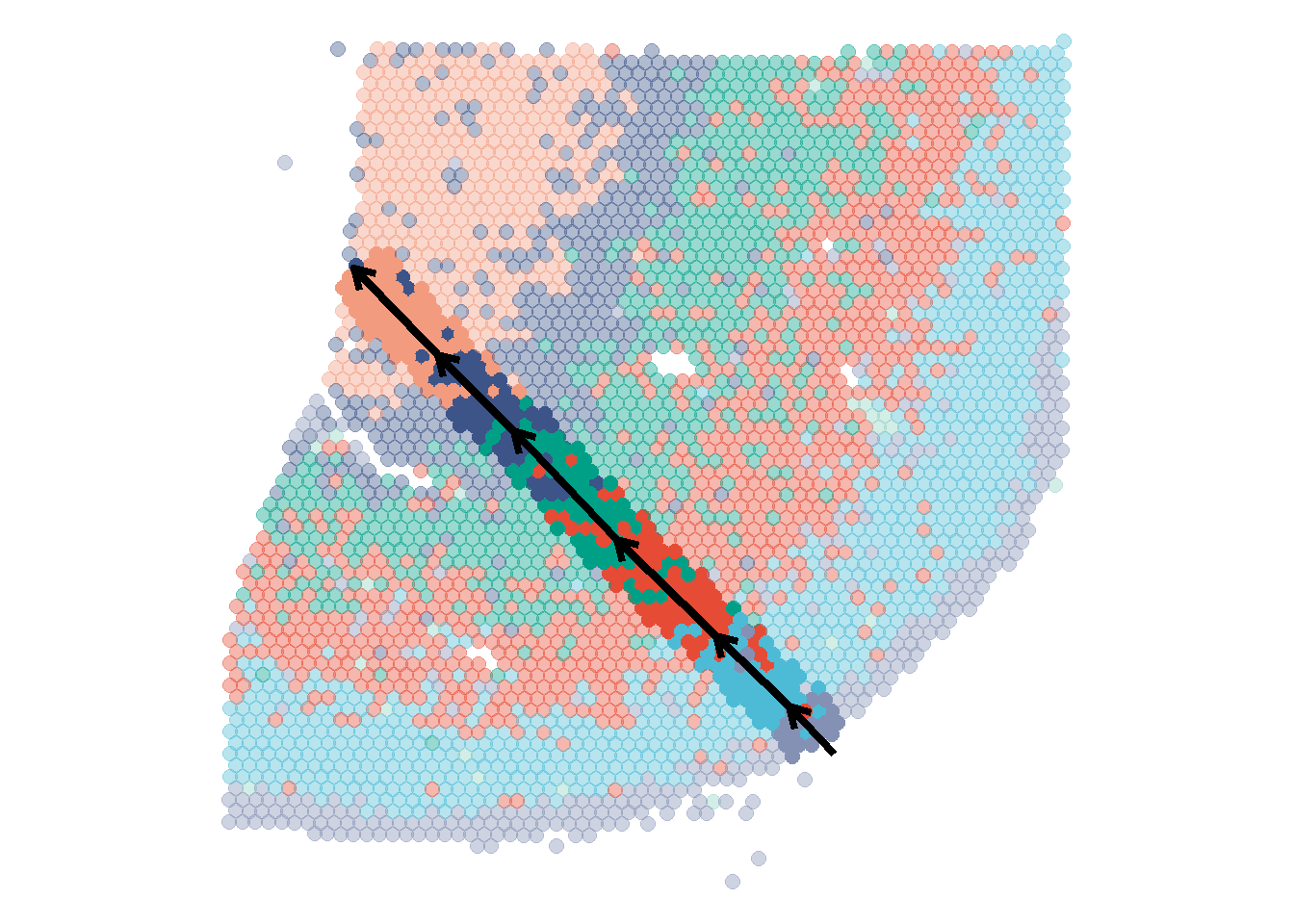
Figure 2.1 A trajectory drawn through all cortical layers of the central nervous system
Let’s try and give our data a directional component using basic functions of the tidyverse.
1. Extracting data
At first we need to extract the respective data from our spata-object and join it with the information we need. A compiled trajectory data.frame contains information about the position of barcode-spots with respect to the trajectory of interest.
cns_trajectory <- getTrajectoryObject(spata_obj_cns, trajectory_name = "cns-layers") compiled_trajectory_df <- cns_trajectory@compiled_trajectory_df # output compiled_trajectory_df
2. Join with information
We are interested to work with the seurat-clusters information and we therefore need to join our data.frame with this variable. Although compiled_trajectory_df already contains variables (whoose meaning we explain down below) it still contains the variables barcodes and sample and it is thus a perfectly valid input for argument spata_df of the joinWith()-function.
joined_df <- joinWith(spata_obj_cns, spata_df = compiled_trajectory_df, features = "seurat_clusters") # output joined_df
3. Apply dplyr::mutate()
The variable projection_length refers to the distance between a barcode-spot’s orthogonal projection onto the trajectory and the beginning of the trajectory. Rounding these projection-length values with a given accuracyeffectively bins the barcode-spots in small groups according to their position of the trajectory’s progress. dplyr::mutate() comes in handy to easily create two new variables.
plot_df <- mutate(.data = joined_df, order_binned = plyr::round_any(x = projection_length, accuracy = 5, f = floor), trajectory_order = stringr::str_c(trajectory_part, order_binned, sep = "_")) # output plot_df
4. Plotting with ggplot2
The functions applied did not change the fact that an observation in plot_df still refers to a barcode spot and the variables of the data.frame still refer to the information we have about each barcode-spot.
ggplot(data = plot_df) + geom_bar(mapping = aes(x = trajectory_order, fill = seurat_clusters), position = "fill", width = .9) + scale_color_add_on(aes = "fill", clrp = "npg", variable = "discrete") + theme_minimal() + theme( axis.text.x = element_blank(), axis.ticks.x = element_blank(), axis.line.x = element_line(arrow = arrow(length = unit(0.1, "inches"))), panel.grid = element_blank(), legend.position = "bottom" ) + labs(x = "Trajectory Direction", y = NULL, fill = "Seurat Clusters")
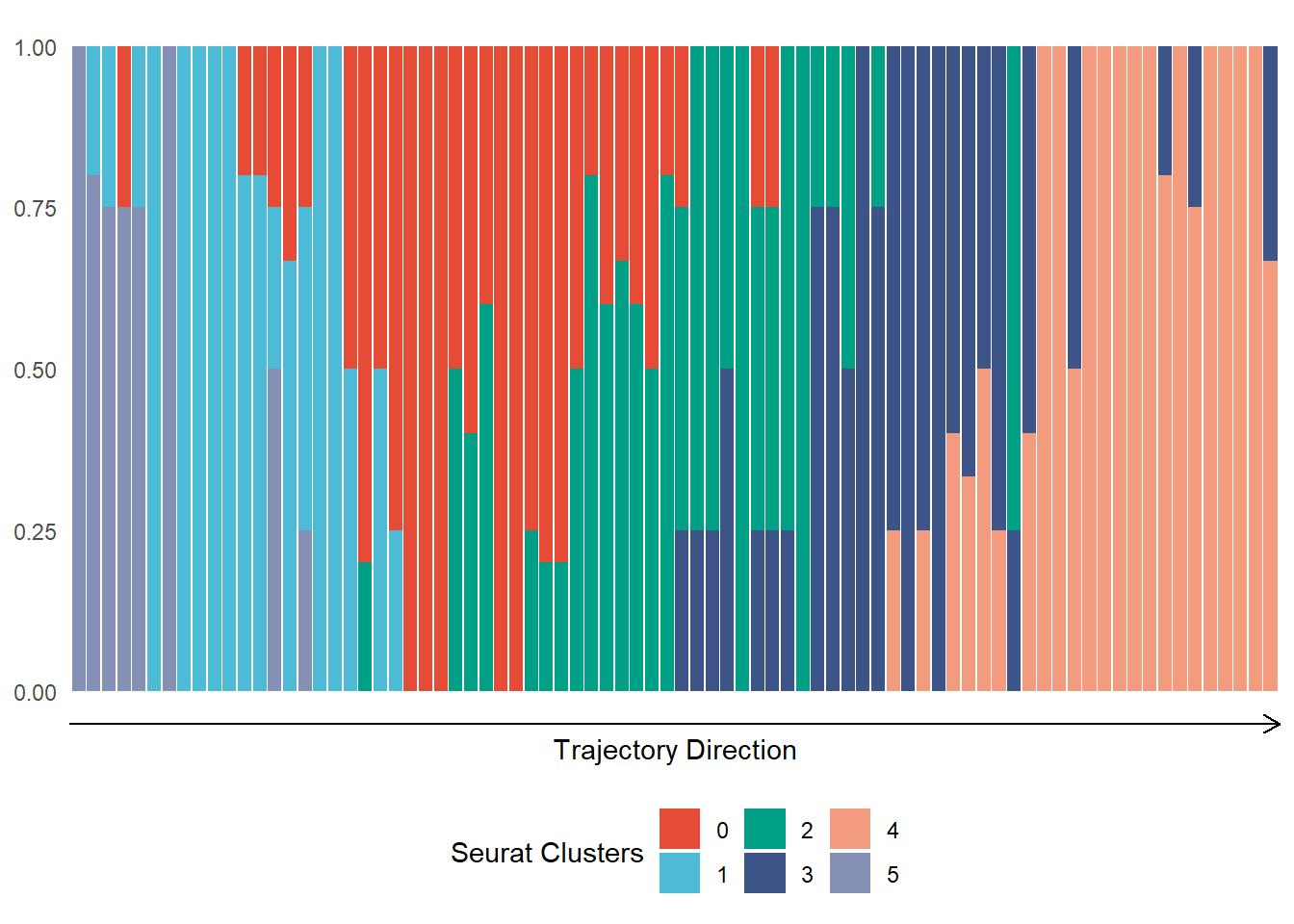
Figure 2.2 Distribution of cluster belonging along the CNS-trajectory
5. Continue analysis and plotting
Once extracted our data.frame can be expanded infinitely. While you can leverage all you programming skills within or outside of the tidyverse SPATA offers a variety of plotting functions that work with extracted, customized data.frames - usually denoted with a 2-suffix.
genes_of_interest <- c("HOPX", "OLIG1", "GFAP", "CCK") df_with_genes <- joinWith(spata_obj_cns, spata_df = plot_df, genes = genes_of_interest) %>% select(-order_binned, -projection_length)#discard currently useless variables plotDistributionAcross2(df = filter(df_with_genes, trajectory_part %in% c("Part 1", "Part 2", "Part 3")), variables = "all", # gene variables are the only numeric variables left across = "seurat_clusters", clrp = "npg") + theme(legend.position = "top") + labs(caption = "First three parts of trajectory") plotDistributionAcross2(df = filter(df_with_genes, trajectory_part %in% c("Part 4", "Part 5", "Part 6")), variables = "all", # gene variables are the only numeric variables left across = "seurat_clusters", clrp = "npg") + theme(legend.position = "top") + labs(caption = "Last three parts of trajectory")

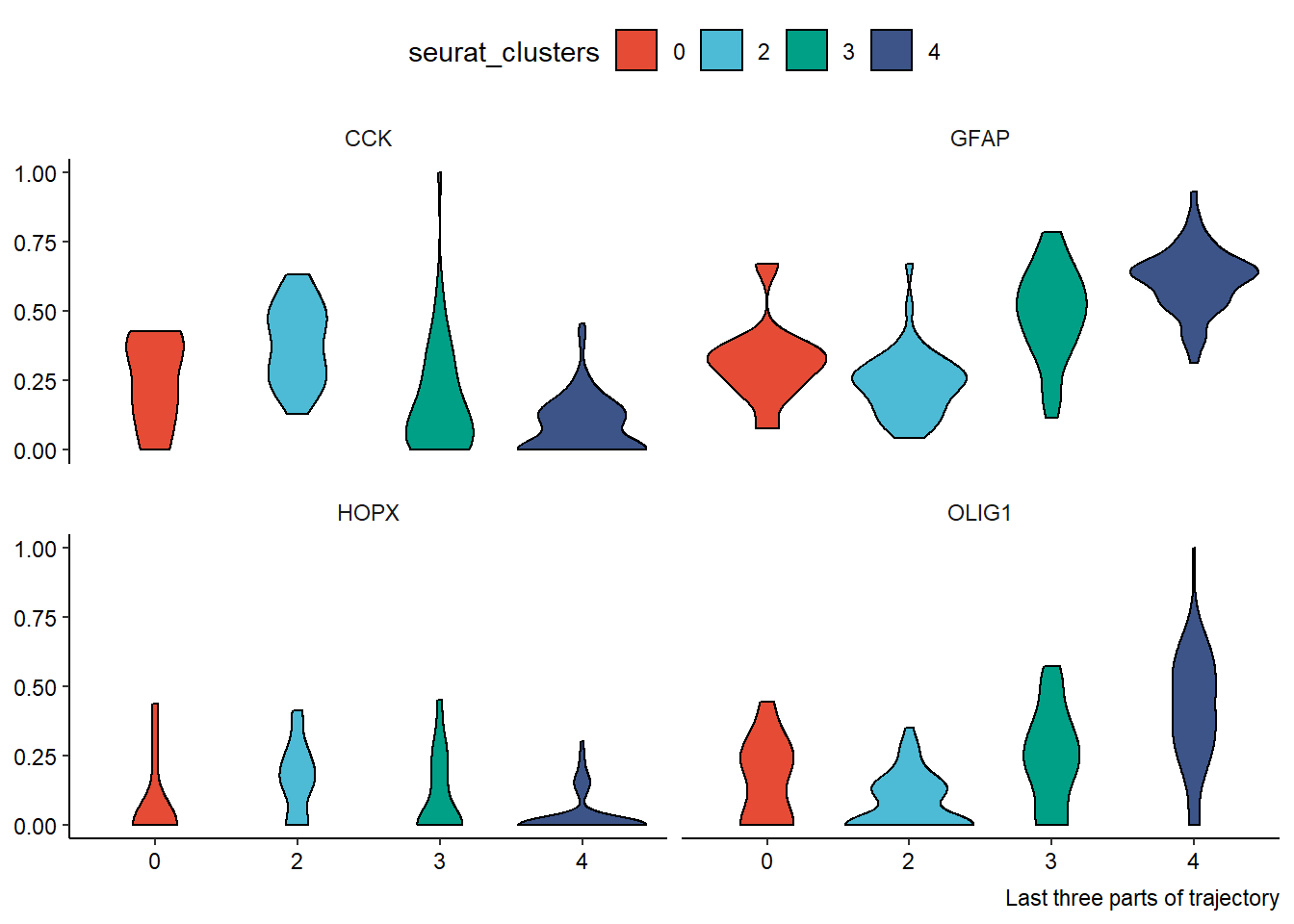
Figure 2.3 Easy application of dplyr’s select() and filter()