Differential Expression Analysis (DEA)
spata-v2-dea.Rmd2. Introduction
Differential expression analysis (DEA) aims to discover quantitative
changes in gene expression levels between defined experimental groups.
Grouping information is stored in form of grouping variables in the
feature data of spata2 object. This includes all
spata-intern generated groups such as spatial segmentation and
clustering as well as any other
grouping variable that has been added via
addFeatures().
library(SPATA2)
library(SPATAData)
library(tidyverse)
# download sample 269_T
object_t269 <- downloadSpataObject(sample_name = "269_T")
object_t269 <-
adjustDefaultInstructions(
object = object_t269,
display_image = TRUE,
clrp = "npg",
pt_clrp = "npg"
)
# download sample 275_T
object_t275 <- downloadSpataObject(sample_name = "275_T")
object_t275 <-
adjustDefaultInstructions(
object = object_t275,
display_image = TRUE,
clrp = "jco",
pt_clrp = "jco"
)
# plot histology images
plotImageGgplot(object = object_t269) +
labs(subtitle = "Sample 269_T")
plotImageGgplot(object = object_t275) +
labs(subtitle = "Sample 275_T")
Fig. 1 The histology of the two example samples.
The grouping variables for this tutorial have been prepared before and can be added like the code shows below.
# load histological/spatial segmentation list
data("spatial_segmentations")
# add histology grouping to spata object
object_t269 <-
addFeatures(
object = object_t269,
feature_df = spatial_segmentations[["269_T"]],
overwrite = TRUE
)
getGroupingOptions(object = object_t269)## factor factor
## "seurat_clusters" "histology"
# load clustering list
data("snn_clustering")
# add cluster grouping to spata object
object_t275 <-
addFeatures(
object = object_t275,
feature_df = snn_clustering[["275_T"]],
overwrite = TRUE
)
getGroupingOptions(object = object_t275)## factor factor factor
## "bayes_space" "seurat_clusters" "snn"Visualize the grouping variables with plotSurface().
plotSurface(
object = object_t269,
color_by = "histology"
)
plotSurface(
object = object_t275,
color_by = "snn"
)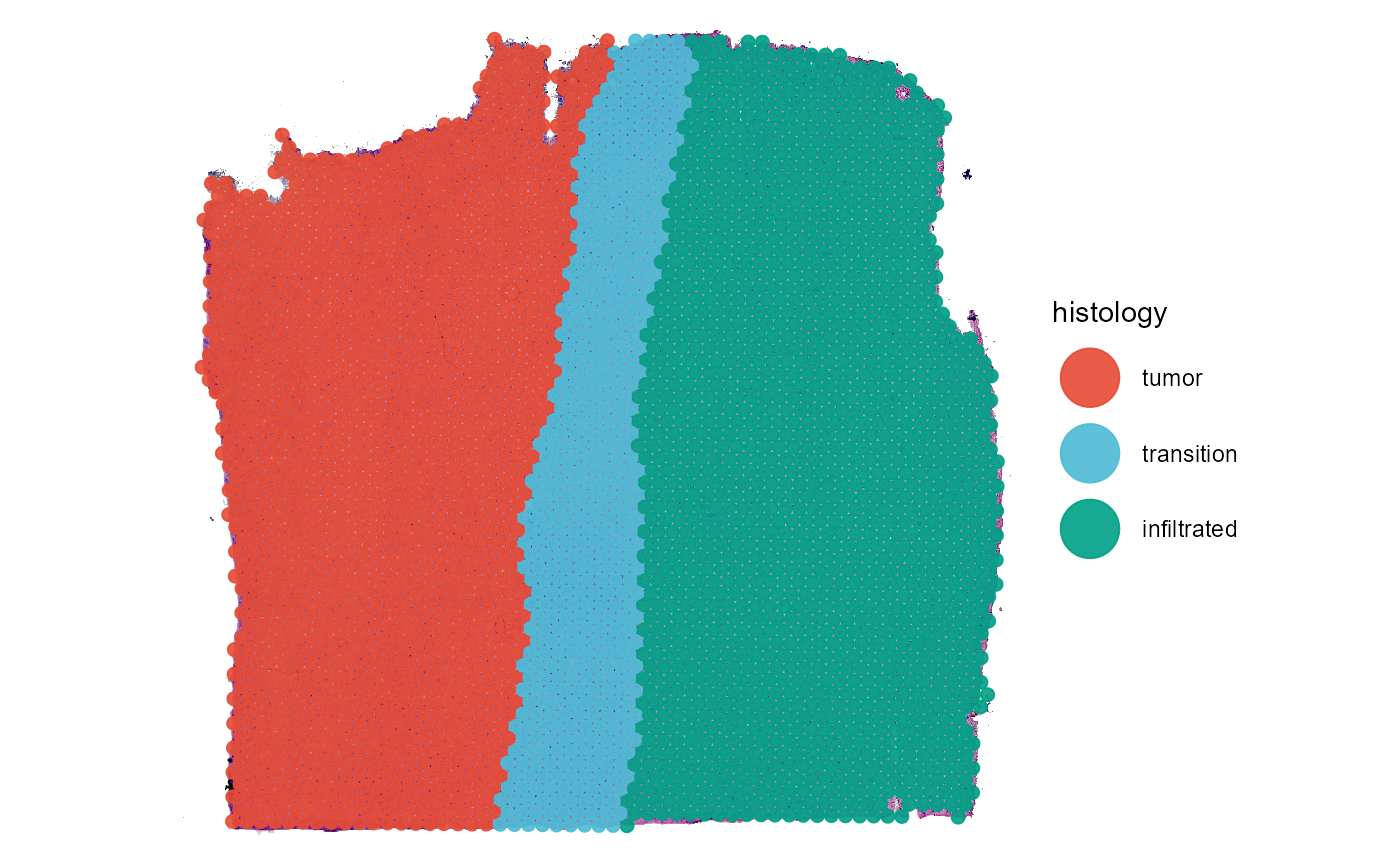
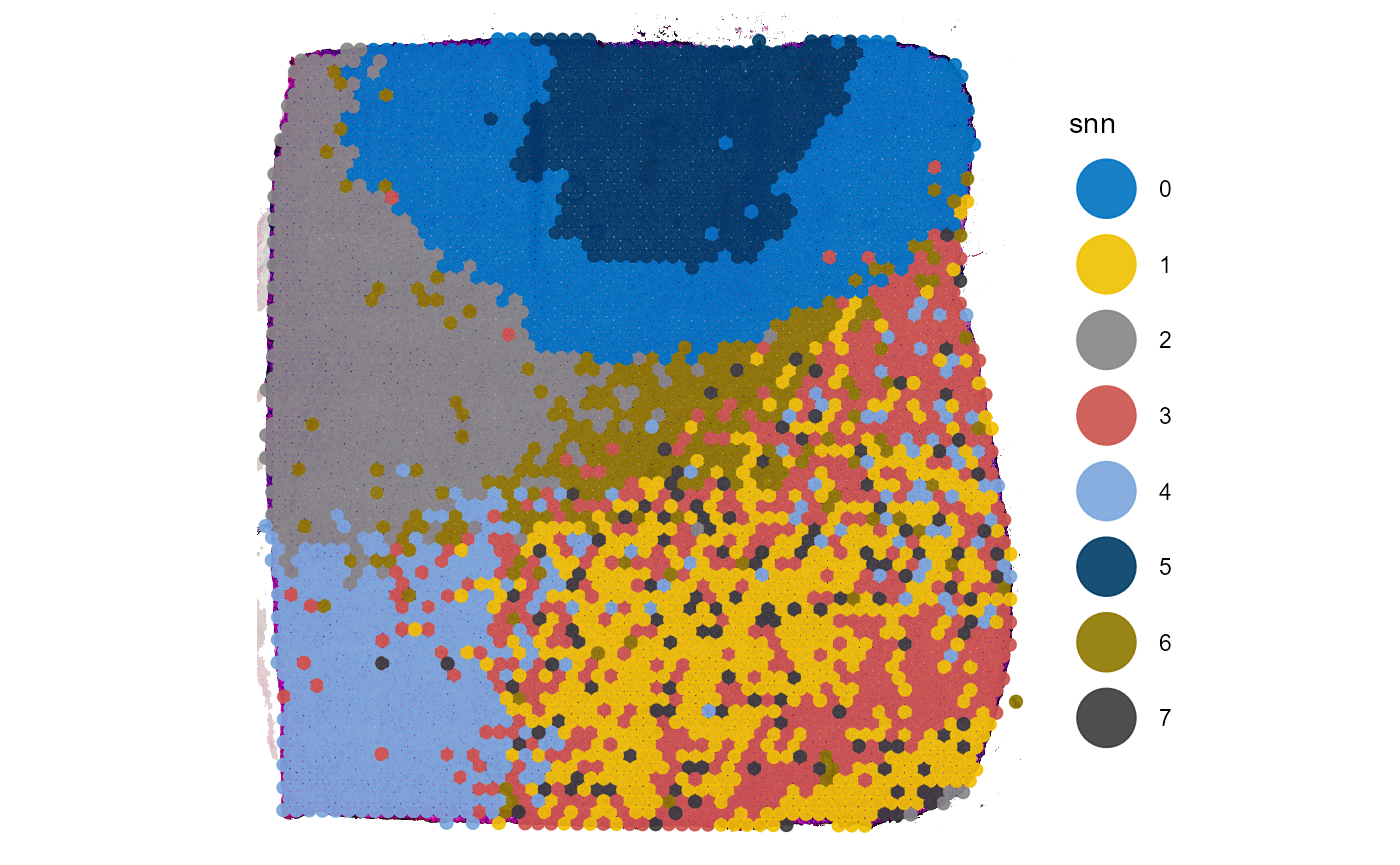
Fig.2 The grouping of the two examples.
3. Running the analysis
DEA in SPATA2 uses the function
Seurat::FindAllMarkers(). It’s output is a data.frame in
which each row corresponds to a gene that turned out to be a marker gene
for one of the identity groups. Additional variables provide information
about it’s p-value, adjusted p-value, logFold change etc.
runDEA() does not alter the output but stores it in the
spata2 object, more precisely in the @dea
slot. With printDeaOverview() you can check across which
groups and according to which method DEA has already been conducted.
object_t269 <- runDEA(object = object_t269, across = "histology", method_de = "wilcox")
printDeaOverview(object = object_t269)## DEA results exist for grouping variables:
##
## - 'seurat_clusters' with methods: wilcox
## - 'histology' with methods: wilcox
object_t275 <- runDEA(object = object_t275, across = "snn", method_de = "wilcox")
printDeaOverview(object = object_t275)## DEA results exist for grouping variables:
##
## - 'snn' with methods: wilcox
## - 'bayes_space' with methods: wilcox3. Extracting results
There are two main functions with which you can manually extract the
DEA results desired. First, getDeaResultsDf() returns the
original resulting data.frame of Seurat::FindAllMarkers().
getDeaGenes() returns a vector of gene names.
# extract the complete data.frame
getDeaResultsDf(
object = object_t275,
across = "snn",
method_de = "wilcox"
)## # A tibble: 3,786 x 7
## p_val avg_log2FC pct.1 pct.2 p_val_adj snn gene
## <dbl> <dbl> <dbl> <dbl> <dbl> <fct> <chr>
## 1 7.77e-182 1.27 0.799 0.277 1.67e-177 0 POSTN
## 2 6.72e-221 1.22 1 0.982 1.44e-216 0 PTN
## 3 1.89e-233 1.22 1 0.996 4.06e-229 0 CST3
## 4 4.55e-180 1.18 0.95 0.583 9.75e-176 0 LANCL2
## 5 1.14e-151 0.997 0.925 0.553 2.44e-147 0 TTYH3
## 6 1.68e-165 0.996 0.995 0.885 3.60e-161 0 HOPX
## 7 2.08e-158 0.983 0.96 0.699 4.47e-154 0 METRN
## 8 3.64e-177 0.980 0.995 0.9 7.80e-173 0 RAMP1
## 9 5.41e-138 0.924 0.994 0.758 1.16e-133 0 RCAN1
## 10 5.57e-157 0.885 0.997 0.751 1.19e-152 0 IGFBP2
## # i 3,776 more rowsUsing the arguments across_subset, min_lfc,
n_highest_lfc, max_adj_pval,
n_lowest_pval the output of the function can be adjusted to
specific questions.
# e.g. top 10 genes for histology area 'tumor'
getDeaResultsDf(
object = object_t269,
across = "histology",
across_subset = "tumor", # the group name(s) of interest,
n_highest_lfc = 10, # top ten genes
max_adj_pval = 0.01 # pval must be lower or equal than 0.01
)## # A tibble: 10 x 7
## p_val avg_log2FC pct.1 pct.2 p_val_adj histology gene
## <dbl> <dbl> <dbl> <dbl> <dbl> <fct> <chr>
## 1 0 2.30 1 0.917 0 tumor CCT6A
## 2 0 2.08 1 0.998 0 tumor SEC61G
## 3 0 2.03 1 0.879 0 tumor NIPSNAP2
## 4 0 1.86 0.999 0.909 0 tumor PTN
## 5 0 1.82 1 0.834 0 tumor CD99
## 6 0 1.79 1 0.977 0 tumor SPP1
## 7 0 1.76 1 0.928 0 tumor PTPRZ1
## 8 0 1.75 1 0.865 0 tumor SPARC
## 9 0 1.72 0.995 0.815 0 tumor APOC1
## 10 0 1.60 1 0.844 0 tumor EGFR
# e.g. all upregulated genes
getDeaResultsDf(
object = object_t275,
across = "snn",
min_lfc = 0.1 # genes with lfc bigger than or equal to 0.1
)## # A tibble: 1,953 x 7
## p_val avg_log2FC pct.1 pct.2 p_val_adj snn gene
## <dbl> <dbl> <dbl> <dbl> <dbl> <fct> <chr>
## 1 7.77e-182 1.27 0.799 0.277 1.67e-177 0 POSTN
## 2 6.72e-221 1.22 1 0.982 1.44e-216 0 PTN
## 3 1.89e-233 1.22 1 0.996 4.06e-229 0 CST3
## 4 4.55e-180 1.18 0.95 0.583 9.75e-176 0 LANCL2
## 5 1.14e-151 0.997 0.925 0.553 2.44e-147 0 TTYH3
## 6 1.68e-165 0.996 0.995 0.885 3.60e-161 0 HOPX
## 7 2.08e-158 0.983 0.96 0.699 4.47e-154 0 METRN
## 8 3.64e-177 0.980 0.995 0.9 7.80e-173 0 RAMP1
## 9 5.41e-138 0.924 0.994 0.758 1.16e-133 0 RCAN1
## 10 5.57e-157 0.885 0.997 0.751 1.19e-152 0 IGFBP2
## # i 1,943 more rows4. Visualize results
4.1 Heatmaps
plotDeaHeatmap() visualizes the results of DEA by using
to the results you would extract with
getDeaResultsDf().
hm <-
plotDeaHeatmap(
object = object_t275,
across = "snn",
across_subset = c("0", "2", "4", "6"),
method_de = "wilcox",
n_highest_lfc = 10,
n_bcsp = 100
)
hm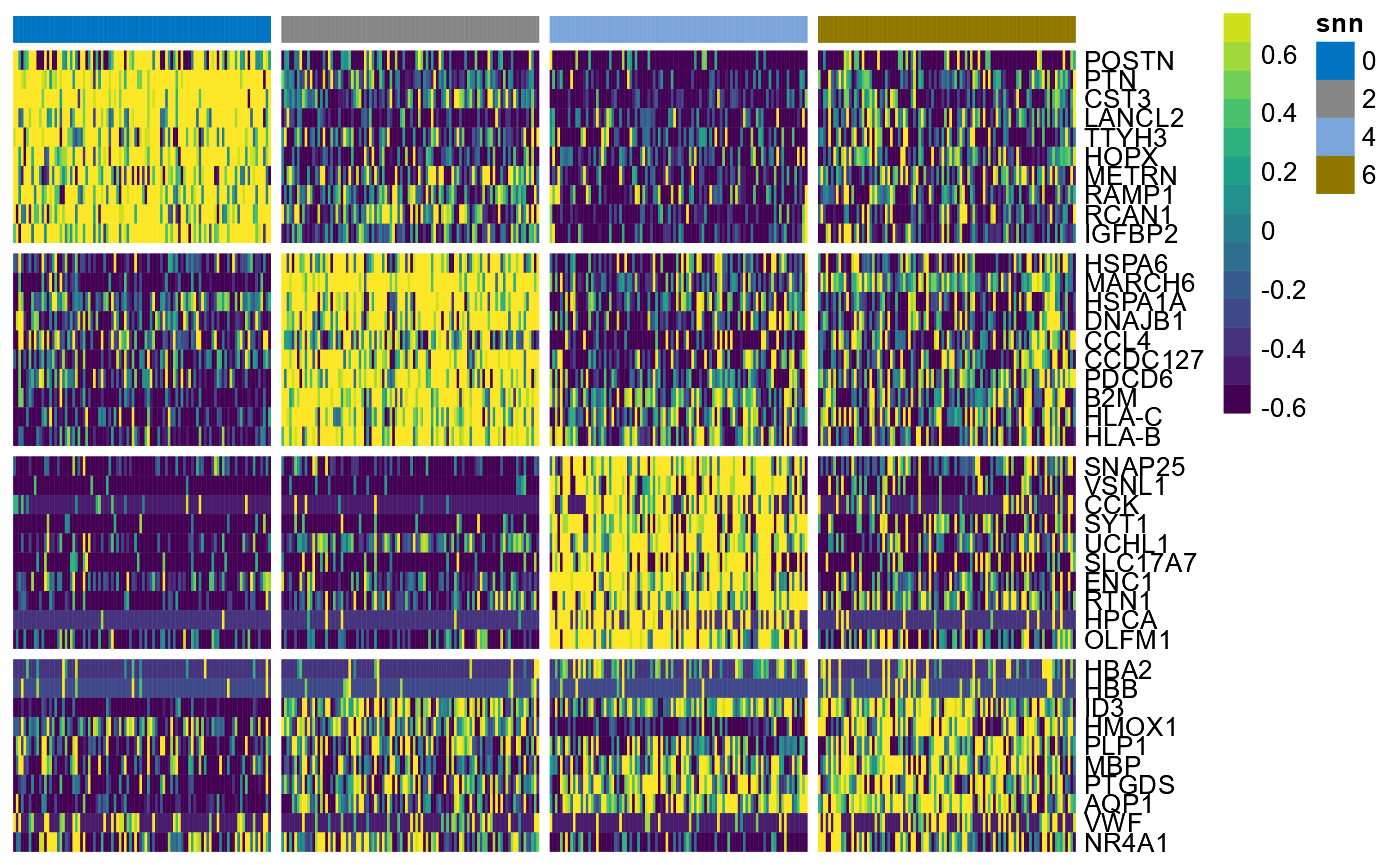
Fig.3 DEA-Heatmap.
4.2 Dotplots
plotDeaDotPlot() visualizes the results of DEA either by
group…
plotDeaDotPlot(
object = object_t275,
across = "snn",
across_subset = c("0", "2", "4"),
n_highest_lfc = 5,
by_group = TRUE,
scales = "free_y",
ncol = 1
)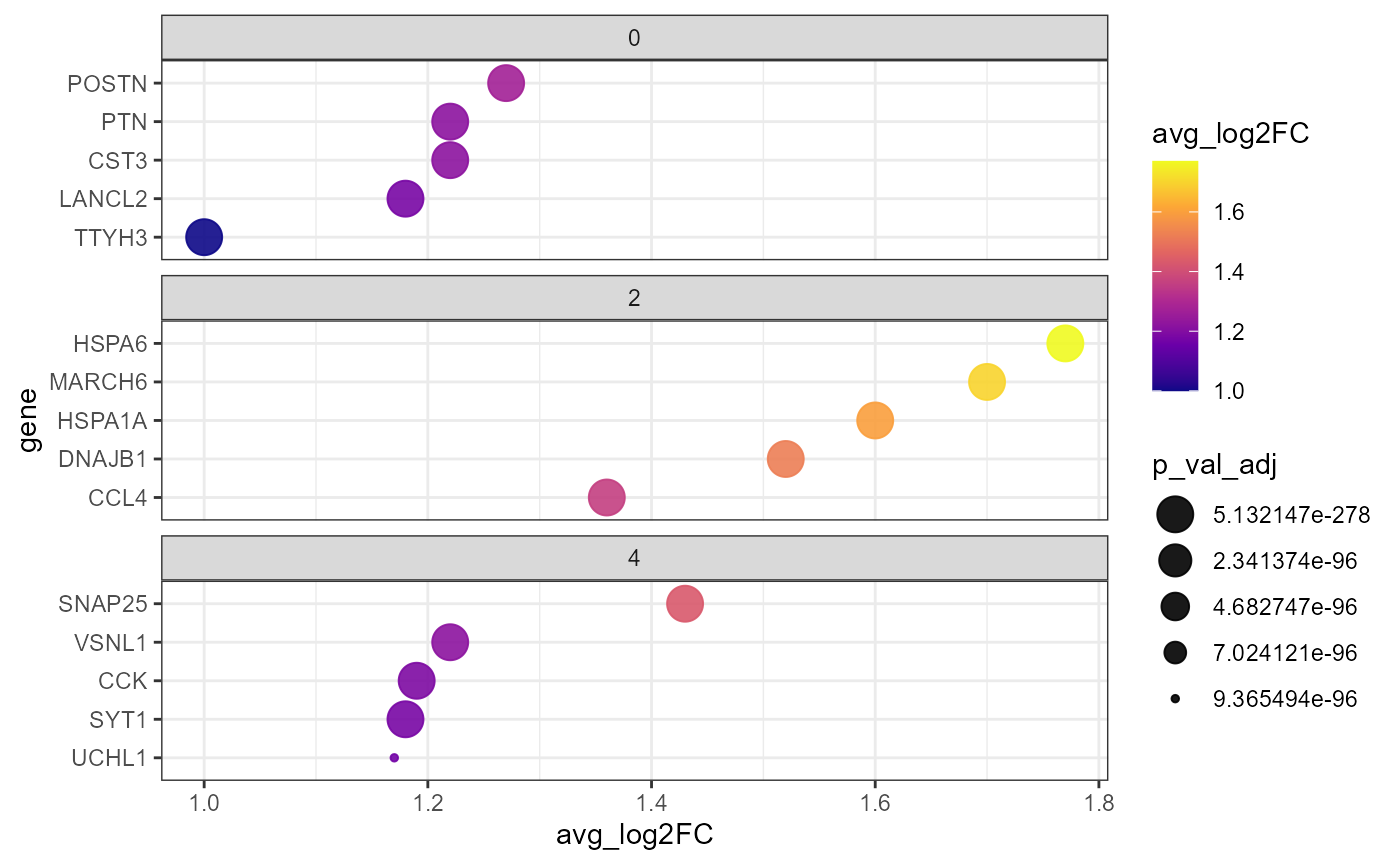
Fig.4 DEA-Dotplot by group.
… or with all groups together.
plotDeaDotPlot(
object = object_t269,
across = "histology",
color_by = "histology",
n_highest_lfc = 5,
by_group = FALSE
)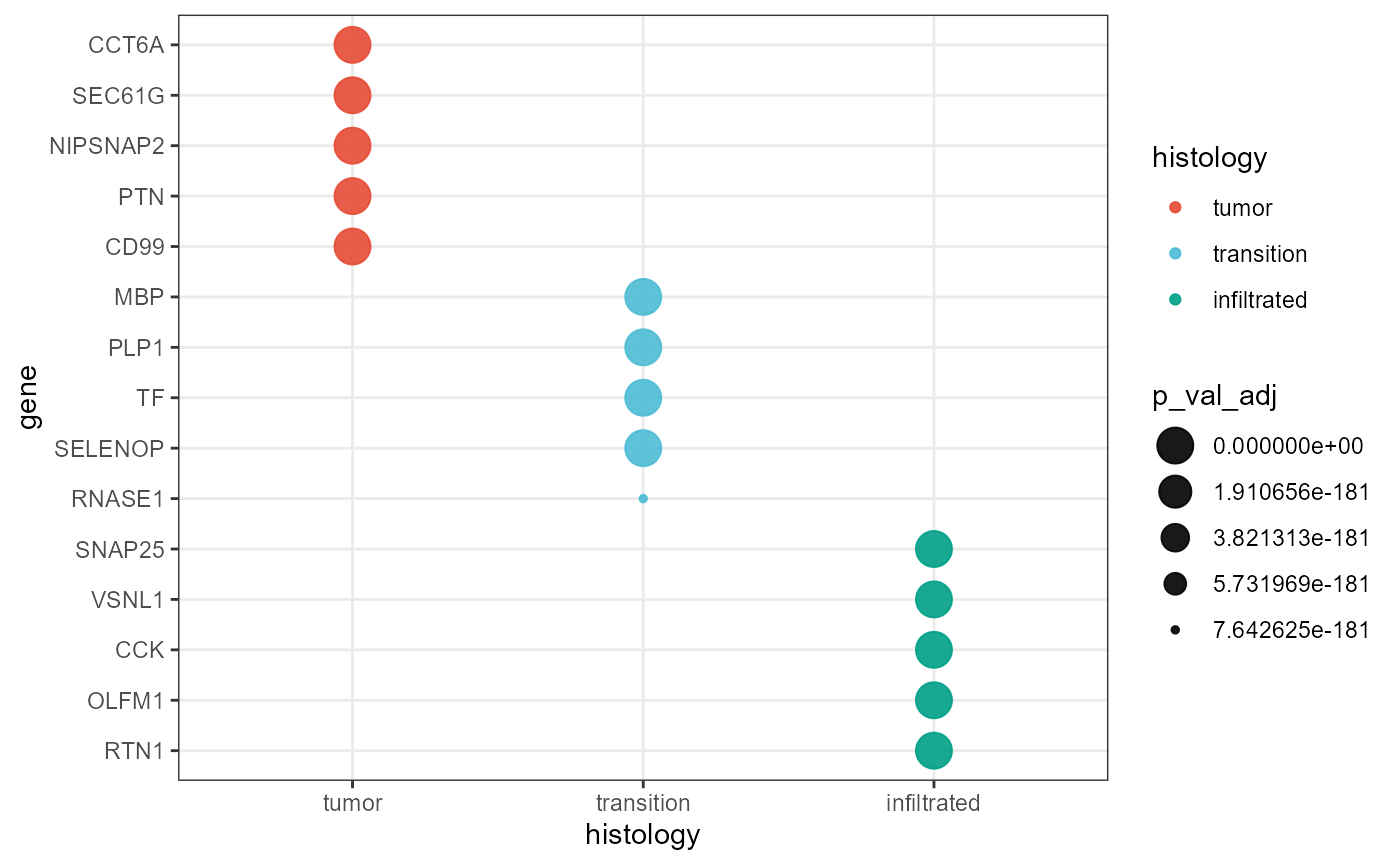
Fig.5 DEA-Dotplot for all groups.
4.3 Box- and Violinplots
There are additional ways to visualize the results of your DEA. As
with almost all plotting functions in SPATA2 a vector
of gene names is necessary for the function to know which genes to plot.
getDeaGenes() is the second function to extract DEA results
and a wrapper around getDeaResultsDf() that returns a
vector gene names.
genes_of_interest <-
getDeaGenes(
object = object_t269,
across = "histology", # the grouping variable
method_de = "wilcox", # the method with which the results were computed
n_highest_lfc = 10,
max_adj_pval = 0.01
)
head(genes_of_interest) # first six## tumor tumor tumor tumor tumor tumor
## "CCT6A" "SEC61G" "NIPSNAP2" "PTN" "CD99" "SPP1"
tail(genes_of_interest) # last six## infiltrated infiltrated infiltrated infiltrated infiltrated infiltrated
## "RTN1" "NPAS4" "SLC1A2" "BEX1" "ENC1" "NRGN"A vector of gene names as returned by getDeaGenes() is a
perfectly valid input for other plotting functions.
top_5_markers_269 <-
getDeaGenes(
object = object_t269,
across = "histology",
n_lowest_pval = 5,
min_lfc = 0.1 # set min_lfc! else downregulated genes are included
)
top_5_markers_269## tumor tumor tumor tumor tumor transition
## "CCT6A" "SEC61G" "NIPSNAP2" "PTN" "CD99" "PLP1"
## transition transition transition transition infiltrated infiltrated
## "MBP" "TF" "SELENOP" "MAG" "SNAP25" "VSNL1"
## infiltrated infiltrated infiltrated
## "CCK" "OLFM1" "RTN1"
# plot results for t269
plotBoxplot(
object = object_t269,
variables = top_5_markers_269,
across = "histology",
nrow = 3
) +
theme(axis.text.x = element_blank(), axis.ticks.x = element_blank()) +
legendTop()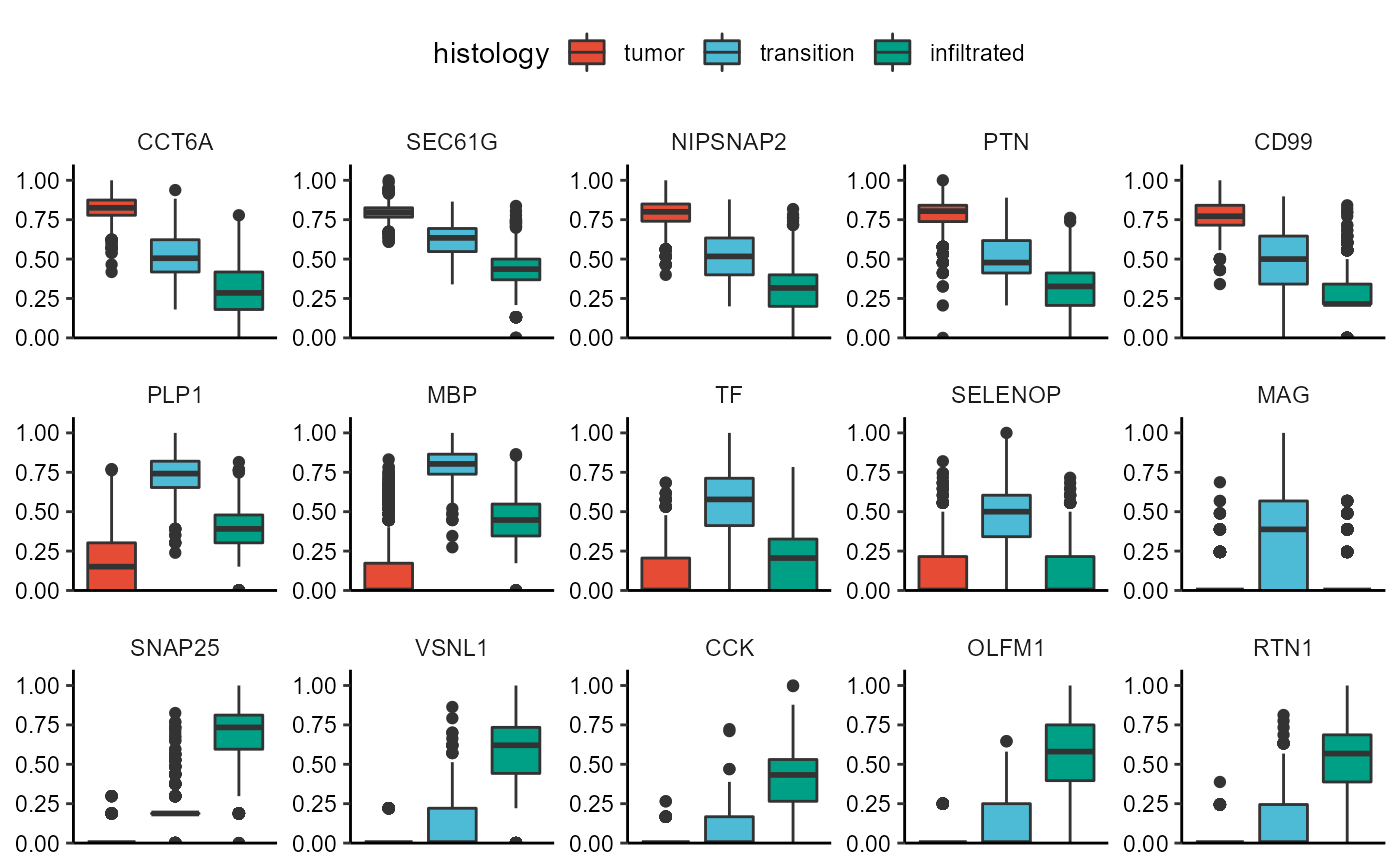
Fig.7 DEA-Boxplots
4.3 Surface plots
Of course, another way to visualize your results are surface plots.
plotSurfaceComparison(
object = object_t269,
color_by = top_5_markers_269 ,
nrow = 3
) +
legendNone()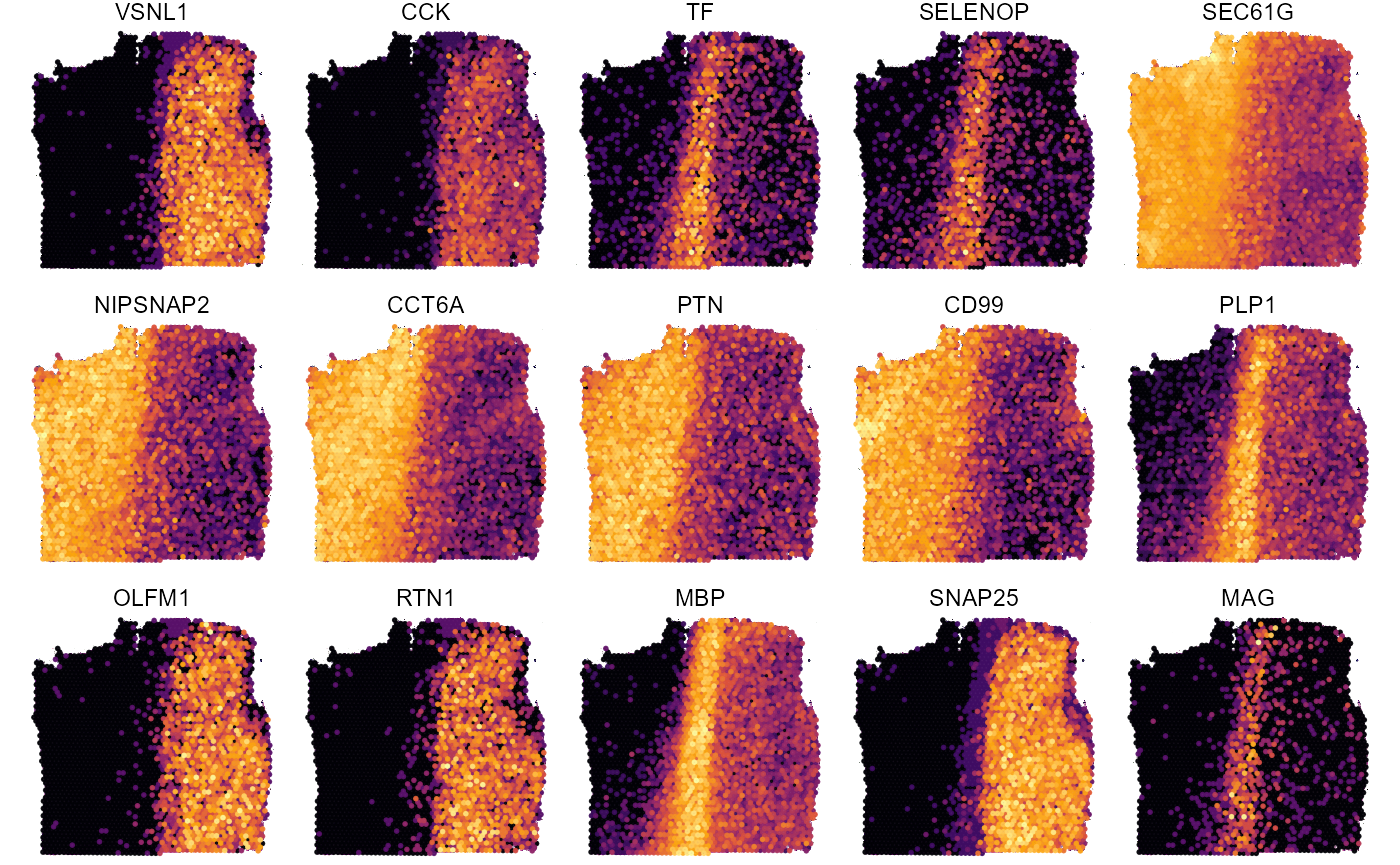
Fig.8 Surface plots with DEA results for sample 269_T.
top_5_markers_275 <-
getDeaGenes(
object = object_t275,
across = "snn",
across_subset = c("0", "2", "4"),
n_lowest_pval = 5,
min_lfc = 0.1 # set min_lfc! else downregulated genes are included
)
plotSurfaceComparison(
object = object_t275,
color_by = top_5_markers_275 ,
nrow = 3
) +
legendNone()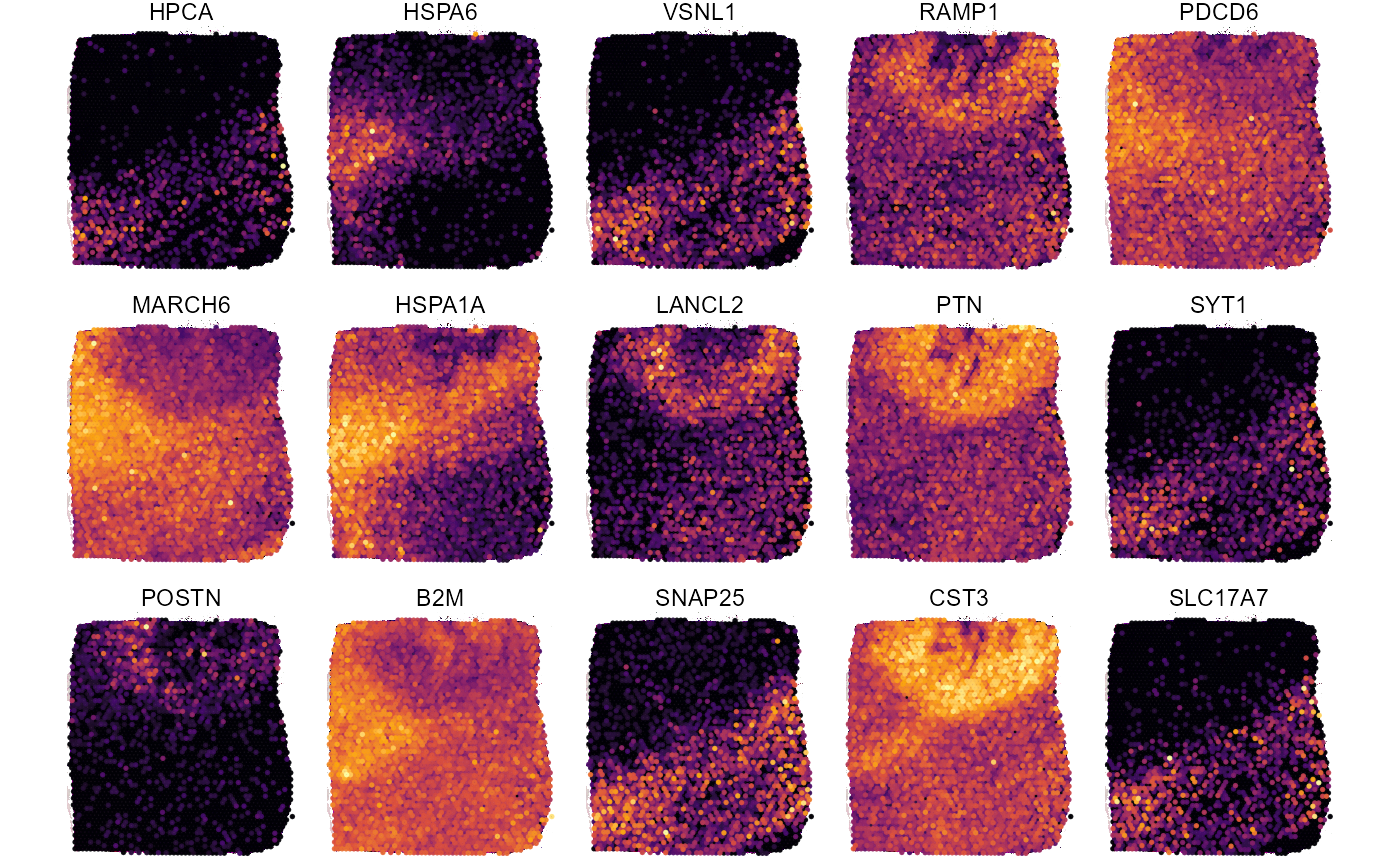
Fig.9 Surface plots with DEA results for sample 275_T.