SPATAData - A database for spatial transcriptomic datasets
spata-v2-spata-data.RmdThe package SPATAData simplifies access to our data base
of spatial transcriptomic samples.
# install the package with...
devtools::install_github("theMILOlab/SPATAData")
# load package
library(SPATAData)1. Interactive downloads
The simplest way to access spatial transcriptomic data sets is by
using the function launchSpataData(). It opens an
interactive application in which all samples that we currently provide
are displayed. You can conveniently look up and filter for
characteristics such as species, organ, pathology, WHO grades in case of
malignancies, organization etc.
# load package
library(SPATAData)
# open the application
launchSpataData()Be patient. It might take a few moments for the app to load. Once the app has loaded you can use the tab ‘Tissue-Organs’ to skim the organs for which spatial transcriptomic datasets are available.
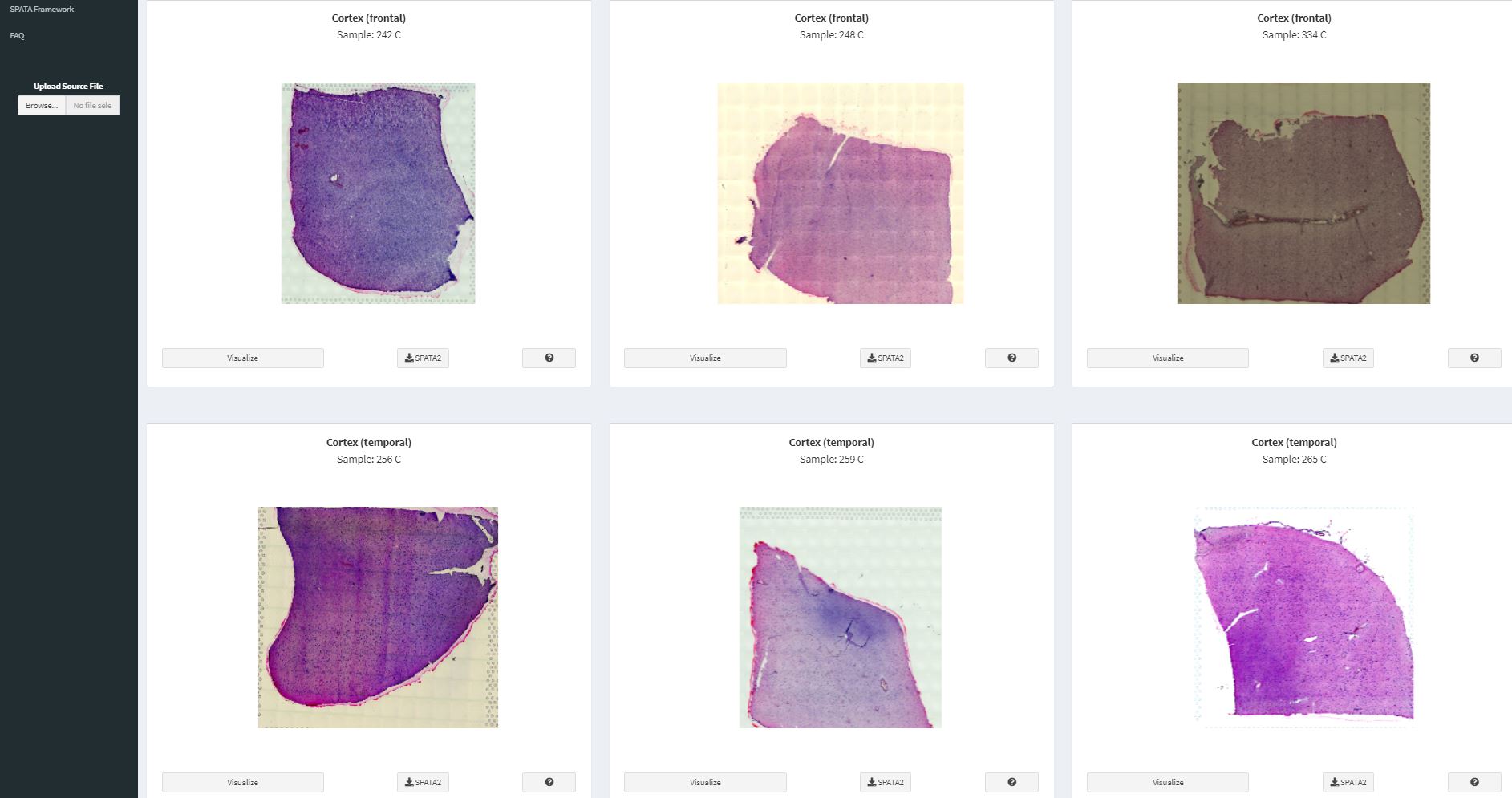
Fig.1 Displaying healthy brain samples that are available.
Using the filter options in the header above you subset all available samples for those that fit the characteristics you are interested in.
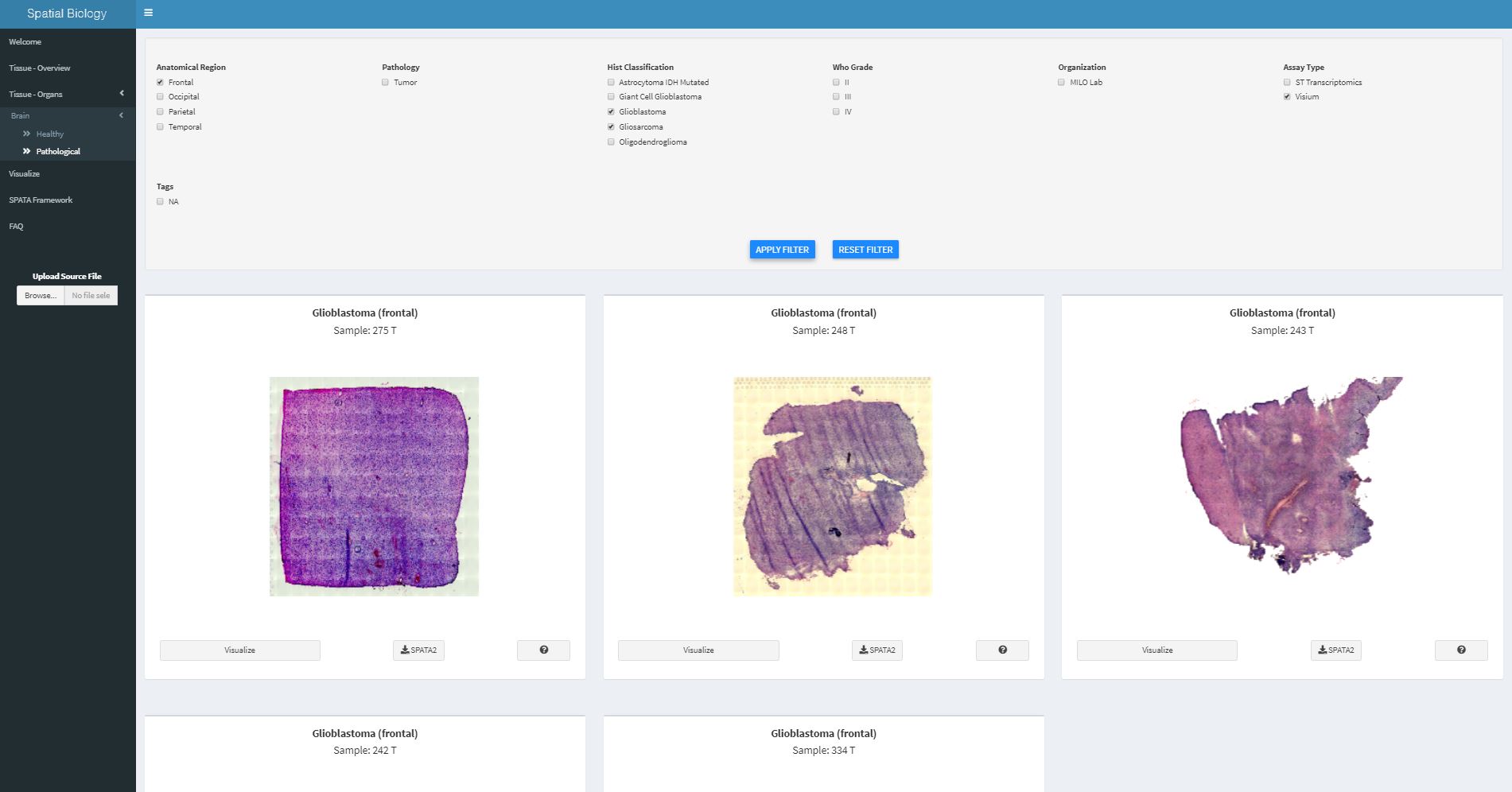
Fig.2 Displaying pathological brain samples that fit the filter characteristics.
Clicking on the button ‘SPATA2’ below every sample downloads the
respective sample in form of a spata2 object. ‘Visualize’
downloads it right in the app where you can visualize its surface in
combination with gene or gene set expression.
2. Manual downloads and other functions
To manually download spata2 objects use the following
functions:
# downloads object and assign it to the gloabl environment
# file = NULL skips saving it
object <- downloadSpataObject(sample_name = "275_T", file = NULL)
# download several samples at the same time
downloadSpataObjects(sample_names = c("275_T", "313_T"), folder = "my_spata_folder")Additional functions:
# obtain all sample names
validSampleNames()
# check how to cite if you have used a sample for your publication
getCitation(object)