Spatial Trajectory Screening
spata-v2-spatial-trajectory-screening.Rmd2. Introduction & overview
Spatial trajectory screening (STS) fits inferred expression changes of genes to predefined models to screen for genes that follow biologically relevant dynamics along a spatial trajectory. This tutorial gives on overview about the most important functions.
Throughout the tutorial we are using the same example sample that we used in the tutorial on spatial trajectories - the glioblastoma sample T269.
As an example we are using a spatial transcriptomic sample of a central nervous system malignancy that features three different, adjacent histological areas: Tumor, a transition zone as well as infiltrated cortex.
library(SPATA2)
library(SPATAData)
library(tidyverse)
object_t269 <- downloadSpataObject(sample_name = "269_T")
# load example image annotations
data("image_annotations")
object_t269 <-
setImageAnnotations(
object = object_t269,
img_anns = image_annotations[["269_T"]],
overwrite = TRUE
)
# plot results
plotImageGgplot(object = object_t269) +
ggpLayerFrameByCoords(object = object_t269) +
ggpLayerThemeCoords()
plotImageAnnotations(
object = object_t269,
tags = "hist_example",
square = TRUE,
expand = 0.5,
encircle = FALSE,
nrow = 2,
display_caption = FALSE
)
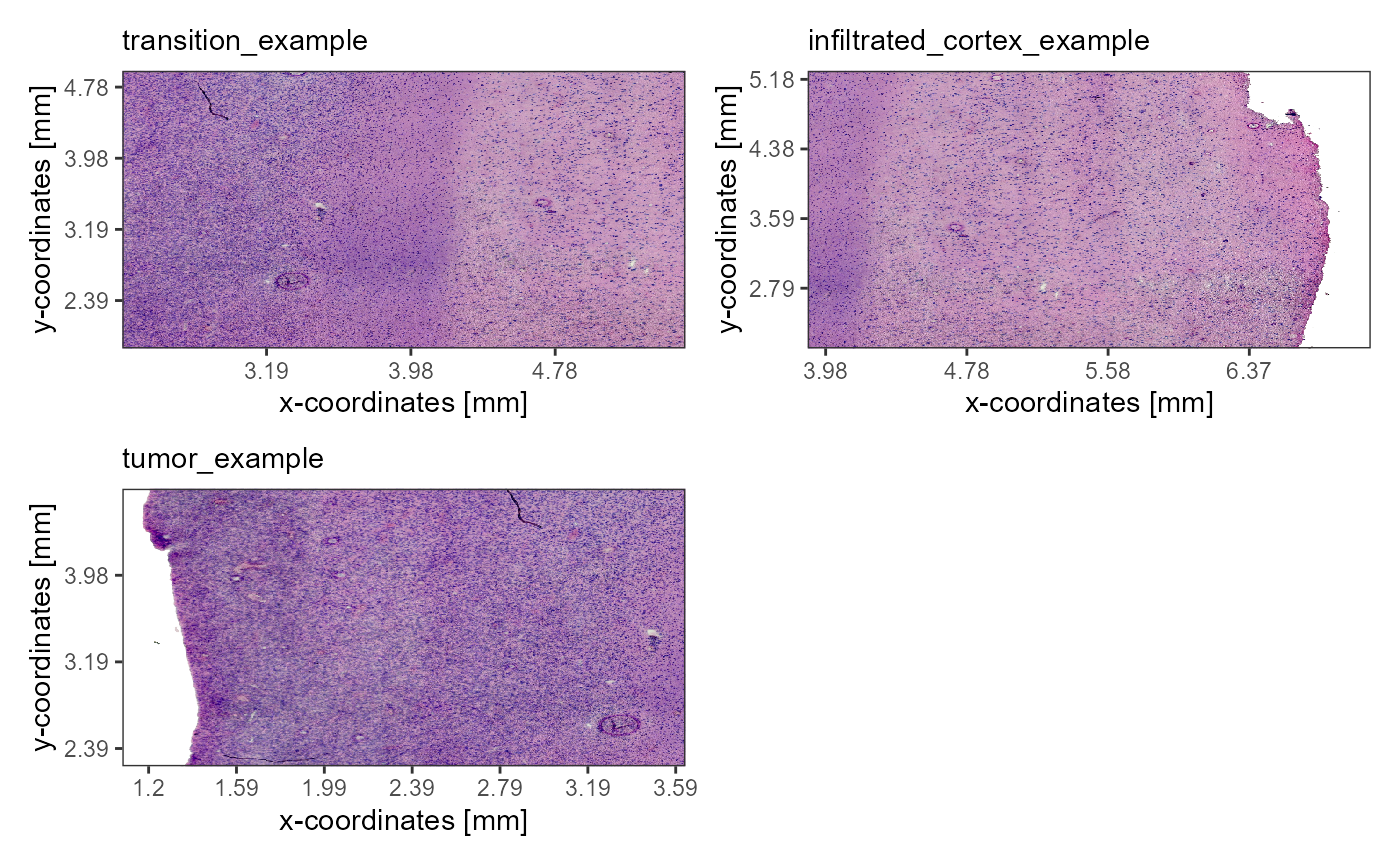
Fig.1 Example sample T269.
The trajectory stays the same, too.
# adds the trajectory that is stored in `spatial_trajectories[["269_T"]][["horizontal_mid"]]`
object_t269 <-
addSpatialTrajectory(
object = object_t269,
id = "horizontal_mid",
start = c("1.5mm", "3.5mm") ,
end = c("6.5mm", "3.5mm") ,
width = "2mm",
overwrite = TRUE
)
plotSpatialTrajectories(
object = object_t269,
ids = "horizontal_mid",
color_by = "MBP"
)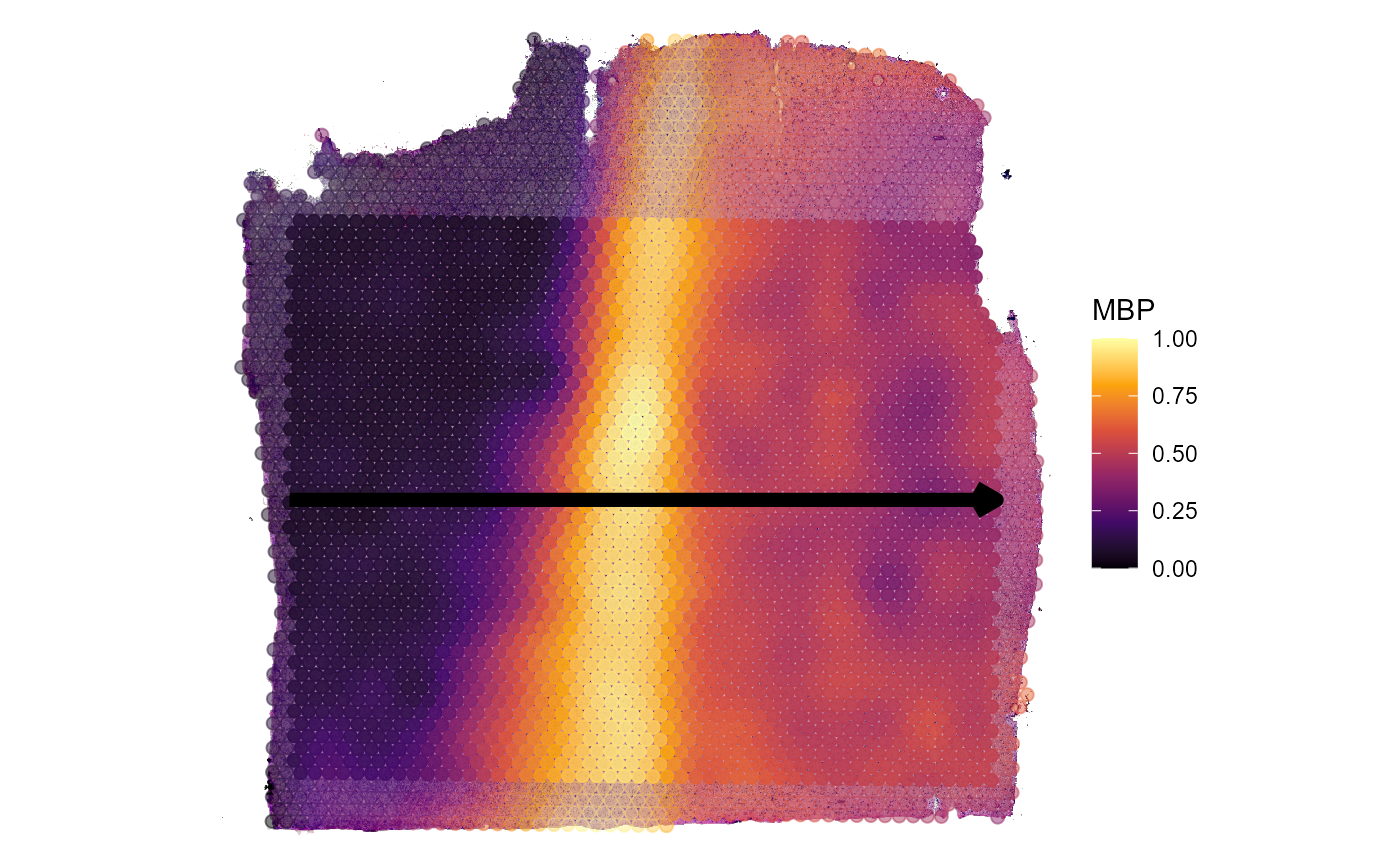
Fig.2 Example spatial trajectory.
3. Run the algorithm
The function to use is called
spatialTrajectoryScreening(). The parameter
variables takes the numeric variables that are supposed to
be included in the screening process. Usually it’s gene names. Spatial
trajectory screening, however, is not restricted to genes as virtually
every numeric variable (e.g. nCount_Genes,
nCount_Features, gene sets, etc.) can be fitted to
spatially meaningful models, too. Therefore, the argument for the input
is simply called variables.
Here, we are using the genes that were already identified as
spatially variable by SPARKX. The goal is to further
analyze which of the genes are expressed in a meaningful way along the
trajectory.
# this is a wrapper around SPARK::sparkx()
object_t269 <- runSparkx(object = object_t269)
spark_df <- getSparkxGeneDf(object = object_t269, threshold_pval = 0.01)
# extract genes with a sparkx pvalue of 0.01 or lower
sparkx_genes <- spark_df[["genes"]]
# show results
spark_df
# show results (> 10 000 genes detected as spatially variable with a p-value of < 0.01)
str(sparkx_genes)## # A tibble: 13,839 x 3
## genes combinedPval adjustedPval
## <chr> <dbl> <dbl>
## 1 ID3 0 0
## 2 MARCKSL1 0 0
## 3 PHC2 0 0
## 4 GNG5 0 0
## 5 CNN3 0 0
## 6 RHOC 0 0
## 7 TXNIP 0 0
## 8 S100A10 0 0
## 9 S100A16 0 0
## 10 C1orf61 0 0
## # i 13,829 more rows## chr [1:13839] "ID3" "MARCKSL1" "PHC2" "GNG5" "CNN3" "RHOC" "TXNIP" ...Once you’ve decided on the input you can run the algorithm.
# catchphrases to subset the models that are of interest (use showModels() to check)
model_subset <- c("sharp_peak", "abrupt_ascending", "abrupt_descending")
STS_T269 <-
spatialTrajectoryScreening(
object = object_t269,
id = "horizontal_mid", # ID of the spatial trajectory
variables = sparkx_genes, # the variables/genes to scree
model_subset = model_subset
)Note: The output of
spatialTrajectoryScreening() is not saved
in the spata2 object but returned in a separate S4 object
of class SpatialTrajectoryScreening. Do
not overwrite the spata2 object by writing
object_t269 <- spatialTrajectoryScreening(object = object_t269, id = "horizontal_mid", ...).
4. Results
4.1 Visualization
To get an overview of the screening as well as a first visualization
of the results use plotOverview(). It sorts genes by their
best model-fit and plots the evaluation score against the p-value for
every model.
plotOverview(
object = STS_T269,
label_vars = 4, # label top 4 variables/genes per model
label_size = 3
)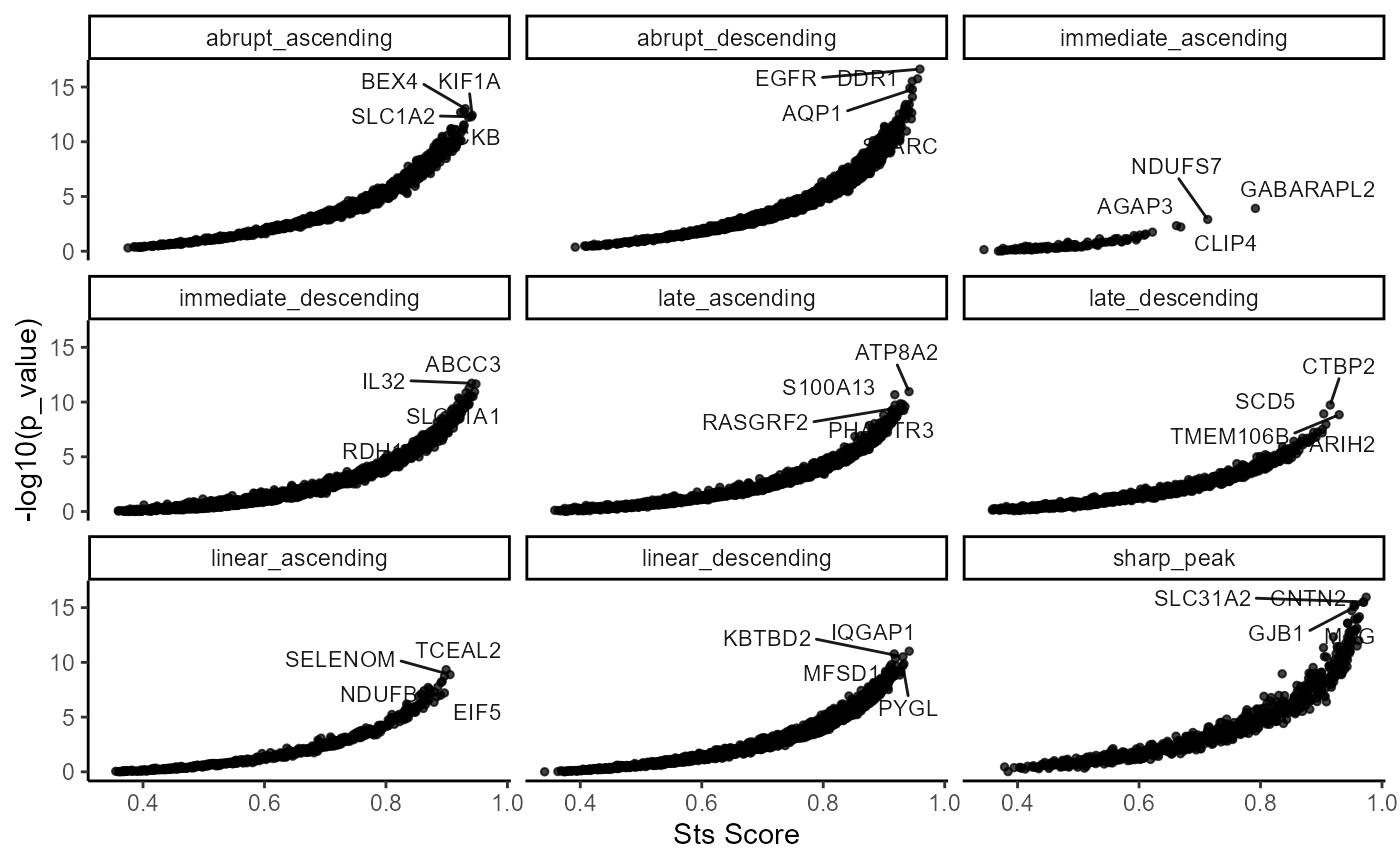
Fig.4 Overview of the screening results.
traj_layer <-
ggpLayerTrajectories(
object = object_t269,
ids = "horizontal_mid",
size = 2.5
)
# abrupt descending
plotSurfaceComparison(
object = object_t269,
color_by = c("EGFR", "DDR1")
) +
traj_layer +
labs(subtitle = "a) Abrupt descending")
# sharp peak
plotSurfaceComparison(
object = object_t269,
color_by = c("CNTN2", "SLC31A2")
) +
traj_layer +
labs(subtitle = "b) Sharp peak")
# abrupt ascending
plotSurfaceComparison(
object = object_t269,
color_by = c("BEX4", "KIF1A")
) +
traj_layer +
labs(subtitle = "c) Abrupt ascending")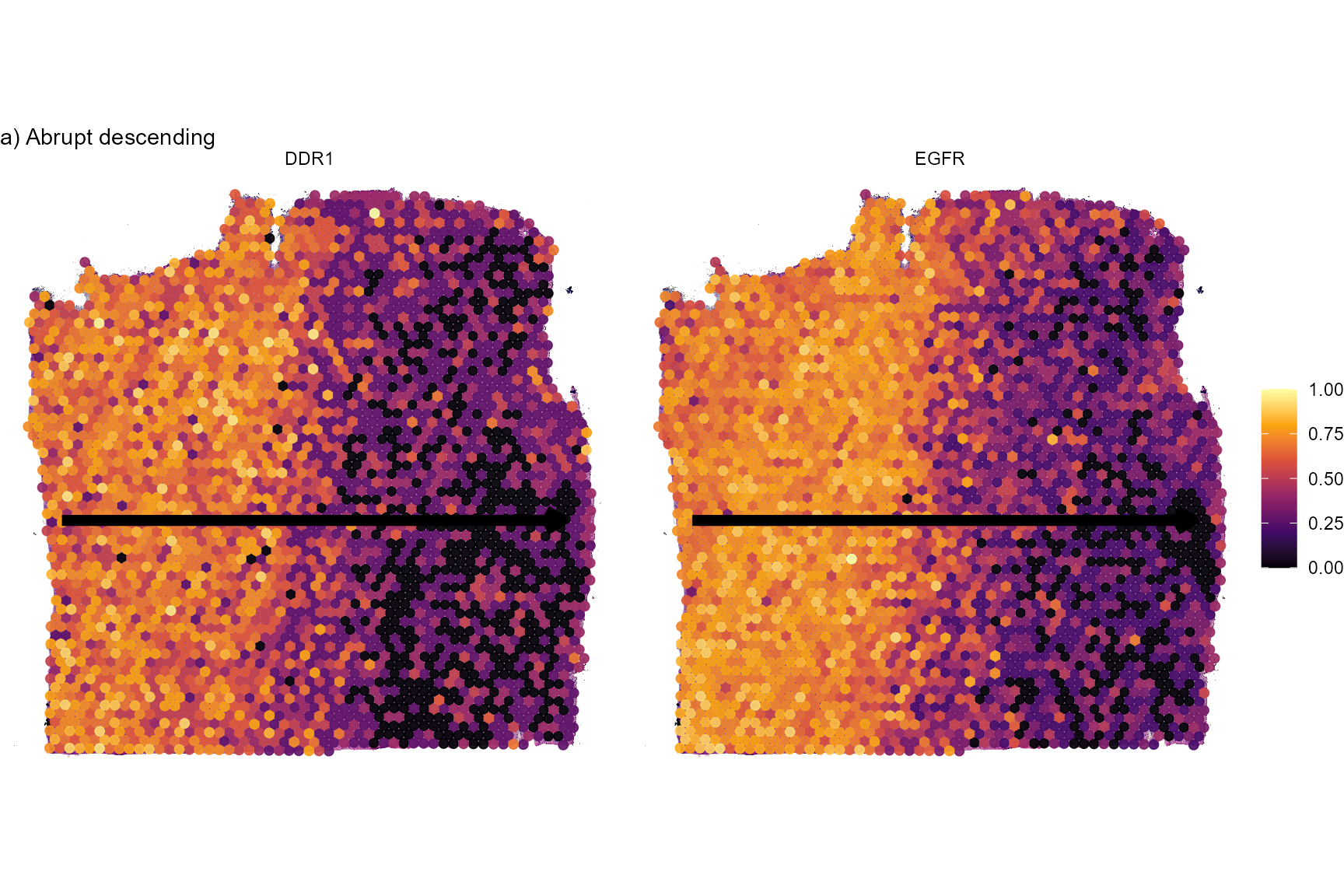
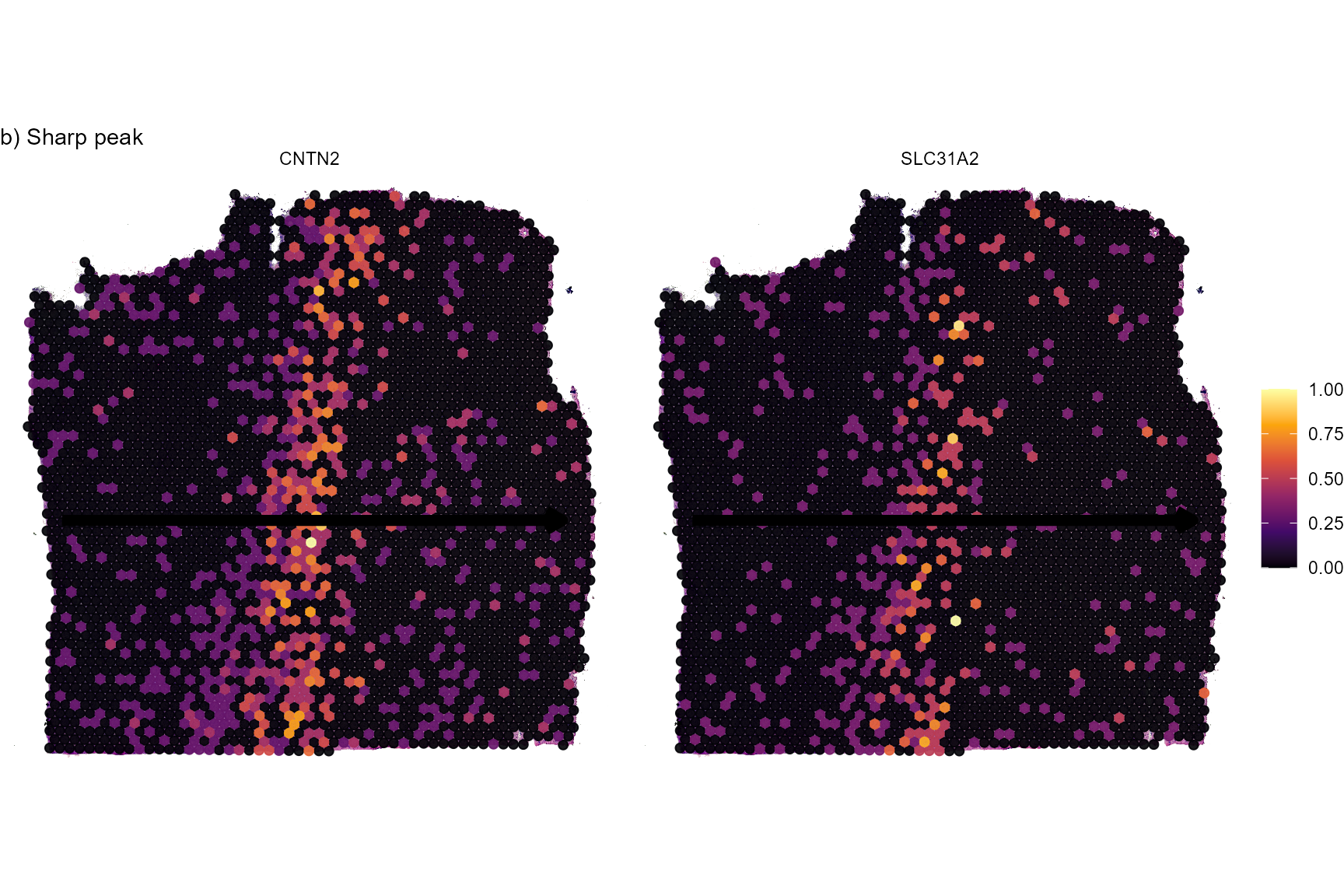
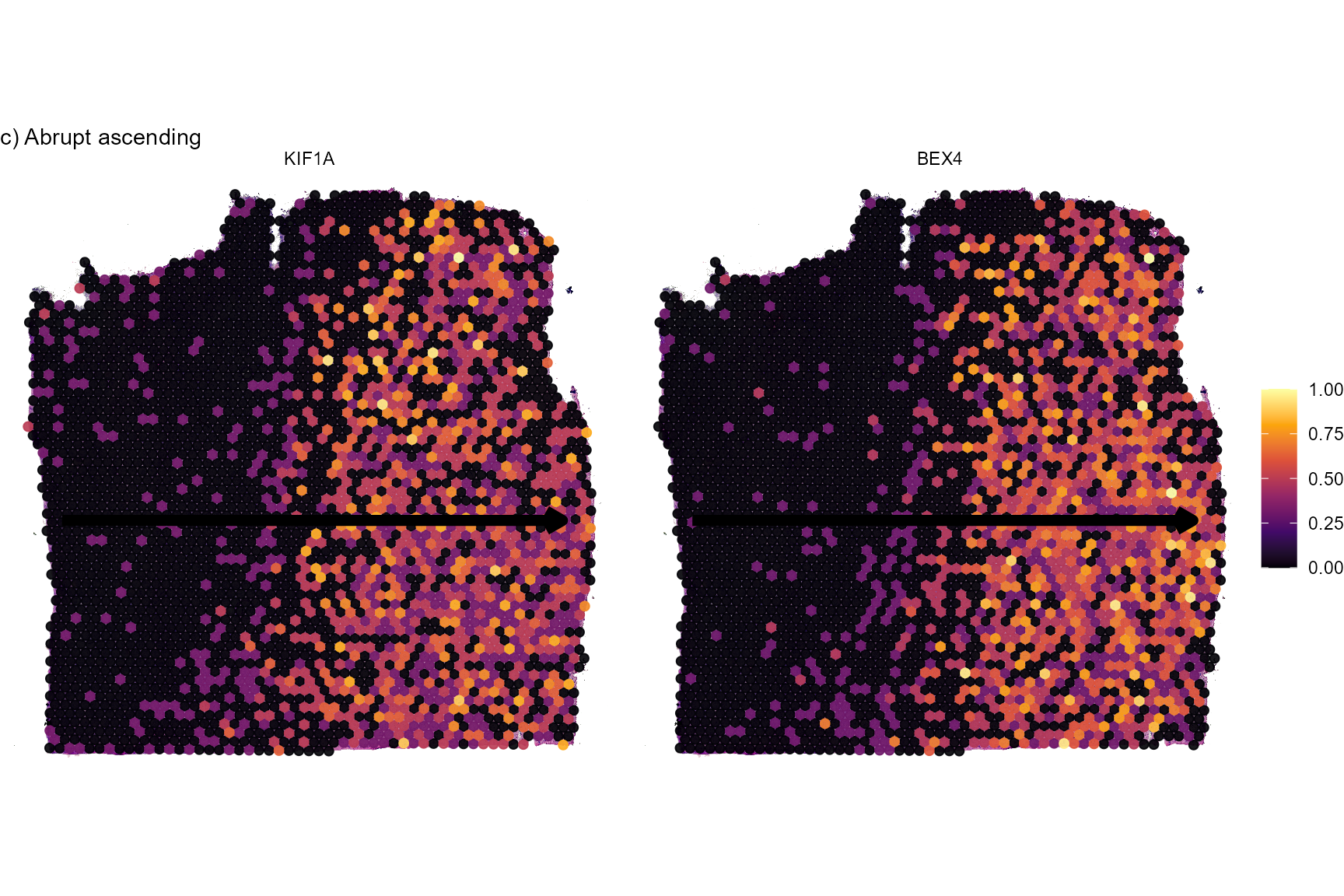
Fig.5 Surface plots of screening results.
The inferred gene expression can be visualized with
plotTrajectoryLineplot().
plotTrajectoryLineplot(
object = object_t269,
id = "horizontal_mid",
variables = c("EGFR", "DDR1", "CNTN2", "SLC31A2", "BEX4", "KIF1A"),
clrp = "sifre",
nrow = 2
)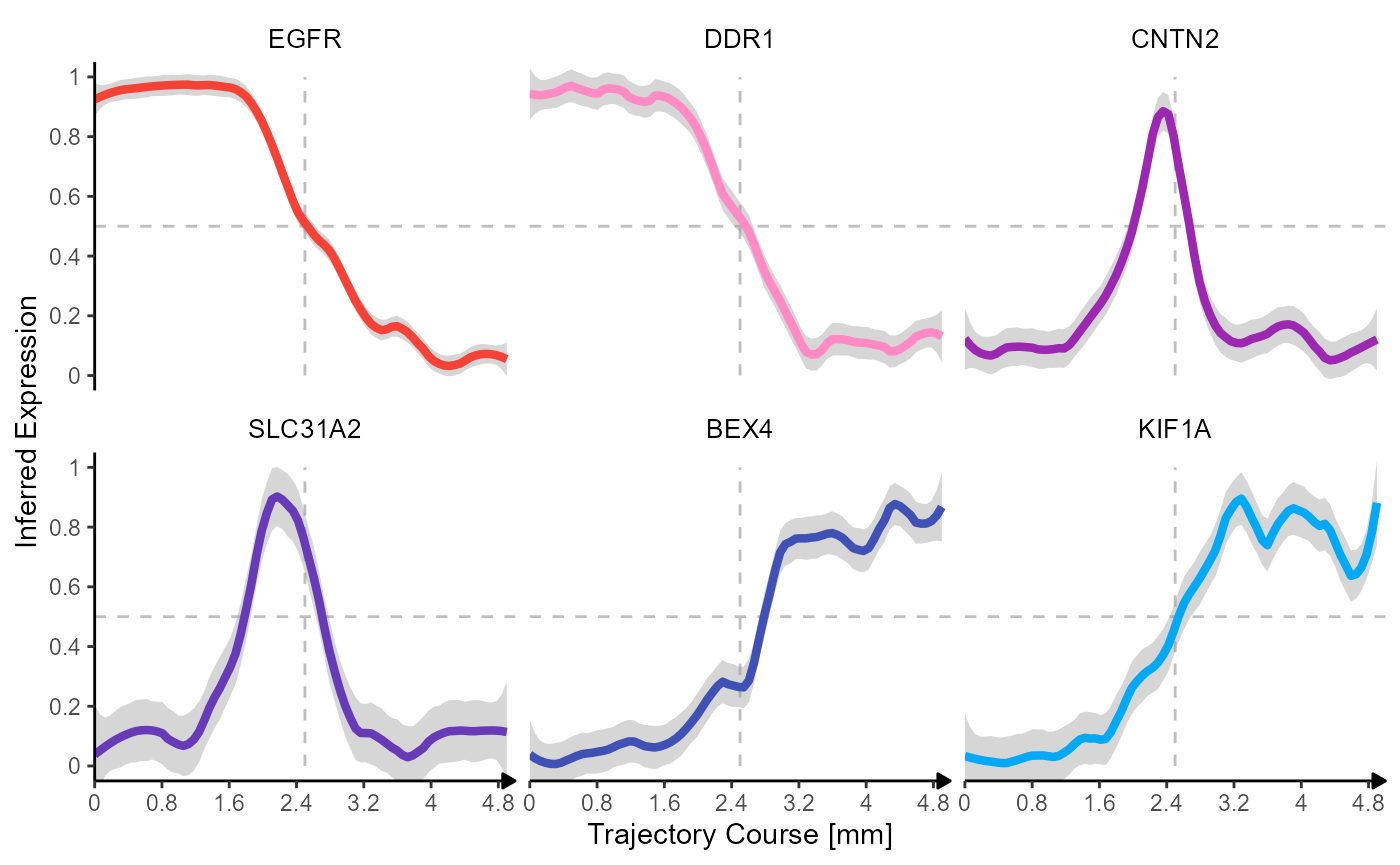
Fig.6 Trajectory lineplots of inferred expression changes.
The used models can always be visualized with
showModels().
showModels(model_subset = model_subset)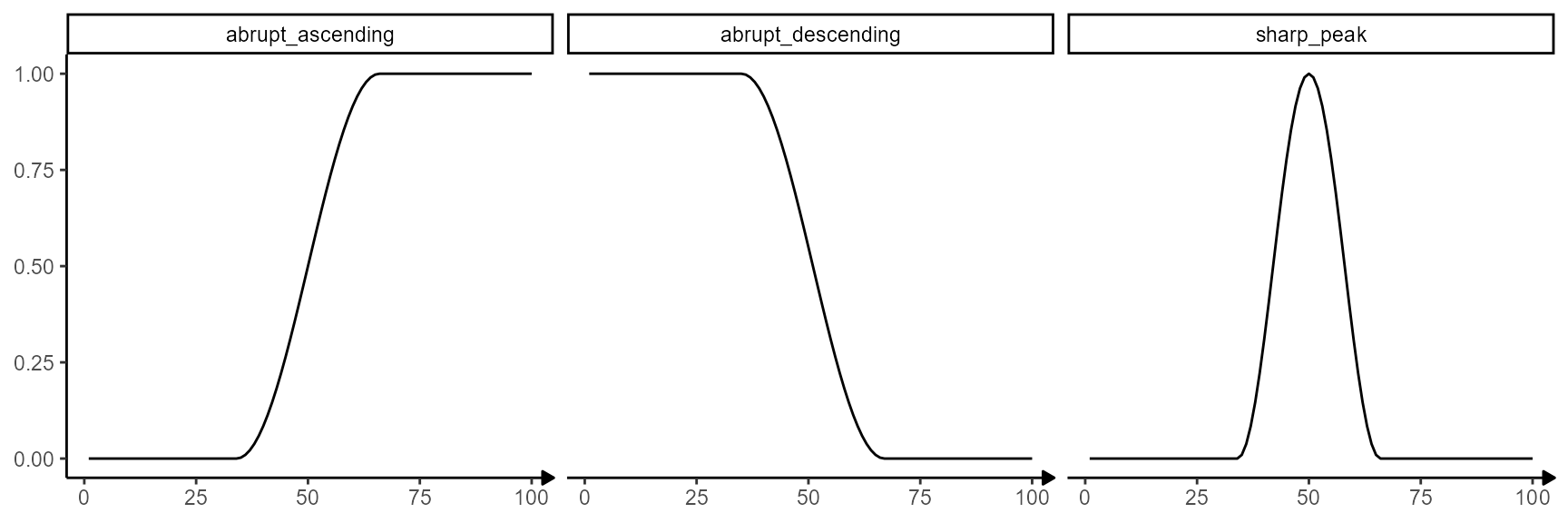
Fig.7 The models that were screened for.
4.2 Extract results
Results can be extracted as data.frames with
getResultsDf() or as a character vector of variable/gene
names with getResultsVec().
res_df <-
getResultsDf(object = STS_T269) %>%
head(200)
res_df## # A tibble: 200 x 8
## # Groups: models [1]
## variables models sts_score corr raoc p_value rauc p_value_adjusted
## <chr> <chr> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl>
## 1 KIF1A abrupt_ascen~ 0.942 0.970 0.914 3.73e-13 1.81 3.44e-10
## 2 SLC1A2 abrupt_ascen~ 0.940 0.969 0.912 5.28e-13 1.85 4.39e-10
## 3 CKB abrupt_ascen~ 0.936 0.969 0.903 5.83e-13 2.04 4.66e-10
## 4 BEX4 abrupt_ascen~ 0.931 0.974 0.887 9.47e-14 2.37 1.27e-10
## 5 ATP1B1 abrupt_ascen~ 0.929 0.962 0.895 3.27e-12 2.21 1.63e- 9
## 6 RTN4 abrupt_ascen~ 0.928 0.973 0.883 1.61e-13 2.45 1.89e-10
## 7 BEX2 abrupt_ascen~ 0.928 0.963 0.893 2.90e-12 2.25 1.52e- 9
## 8 SLC17A7 abrupt_ascen~ 0.927 0.959 0.896 7.33e-12 2.19 2.83e- 9
## 9 RAB11FIP4 abrupt_ascen~ 0.924 0.948 0.900 7.31e-11 2.10 1.56e- 8
## 10 CALM2 abrupt_ascen~ 0.924 0.956 0.891 1.41e-11 2.29 4.54e- 9
## # i 190 more rows
# top 3 peaking genes
peaking_genes <-
getResultsVec(
object = STS_T269,
threshold_eval = 0.5,
threshold_pval = 0.05,
model_subset = "sharp_peak"
) %>%
head(3)
plotSurfaceComparison(
object = object_t269,
color_by = peaking_genes
)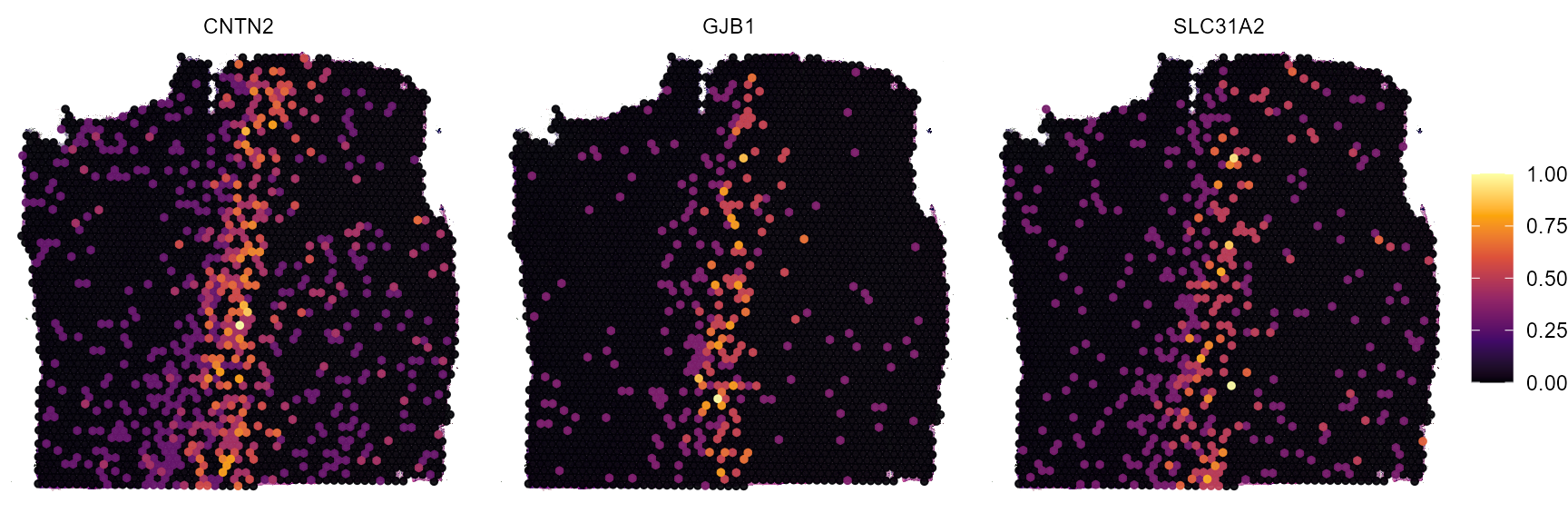
Fig.8 Surface plots of screening results.
5. The S4 SpatialTrajectoryScreening class
Spatial trajectory screening results are provided in an S4 object of
class SpatialTrajectoryScreening. Use
?SpatialTrajectoryScreening to read the description. Note
that SpatialTrajectoryScreening is the class name.
spatialTrajectoryScreening() is the function that runs
the algorithm.
class(STS_T269)## [1] "SpatialTrajectoryScreening"
## attr(,"package")
## [1] "SPATA2"
slotNames(STS_T269)## [1] "binwidth" "coords" "id"
## [4] "method_padj" "models" "n_bins"
## [7] "results" "sample" "summarize_with"
## [10] "spatial_trajectory"